Medullary cystic kidney disease
| Medullary cystic kidney disease | |
|---|---|
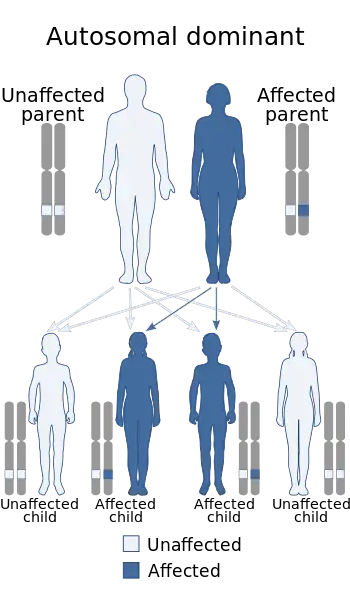 | |
| Medullary cystic kidney disease has an autosomal dominant pattern of inheritance | |
| Specialty | Medical genetics |
| Symptoms | Polydipsia[1] |
| Types | MCKD1 and MCKD2[2][3] |
| Diagnostic method | Kidney biopsy, Kidney ultrasound, CBC[4] |
| Medication | Currently no cure, Drink plenty of fluids, Salt supplement[4] |
Medullary cystic kidney disease (MCKD) is an autosomal dominant kidney disorder characterized by tubulointerstitial sclerosis leading to end-stage renal disease. Because the presence of cysts is neither an early nor a typical diagnostic feature of the disease, and because at least 4 different gene mutations may give rise to the condition, the name autosomal dominant tubulointerstitial kidney disease (ADTKD) has been proposed, to be appended with the underlying genetic variant for a particular individual.[5][6] Importantly, if cysts are found in the medullary collecting ducts they can result in a shrunken kidney, unlike that of polycystic kidney disease. There are two known forms of medullary cystic kidney disease, mucin-1 kidney disease 1 (MKD1) and mucin-2 kidney disease/uromodulin kidney disease (MKD2).[1] A third form of the disease occurs due to mutations in the gene encoding renin (ADTKD-REN), and has formerly been known as familial juvenile hyperuricemic nephropathy type 2.[7]
Signs and symptoms
In terms of the signs/symptoms of medullary cystic kidney disease, the disease is not easy to diagnose and is uncommon. In this condition, loss of kidney function occurs slowly over time, however the following signs/symptoms could be observed in an affected individual:[1]
- Polydipsia
- Enuresis
- Weakness
- Lack of appetite
- Pruritus
- Bone pain
- Pallor
- Nausea
Some individuals with this disease develop gout,[8] which is a condition in which patients develop severe pain and swelling in the big toe or another joint such as the knee. If untreated, it becomes chronic and affects the joints most of the time, instead of intermittently.[9]
Cause
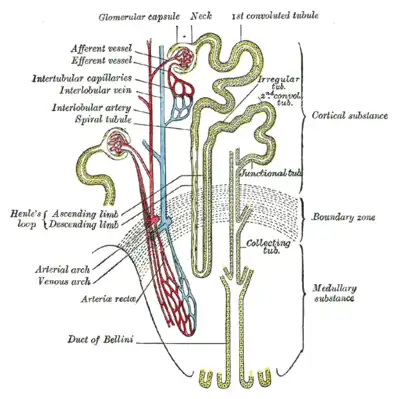
Normal individuals have two copies of the MUC1 and UMOD genes. The genes produce the protein mucin-1 and uromodulin, respectively. These proteins are expressed only in certain cells in the kidney – the thick ascending limb of Henle and distal convoluted tubule – both parts of the kidney tubule. The protein coats the surface of the tubule and protects the tubule.[2][3][1]In MKD, individuals have one normal and one abnormal MUC1 gene. In uromodulin kidney disease (UKD), individuals have one normal and one abnormal UMOD gene. The abnormal (mutated) gene produces a misfolded protein product that deposits within cells of the renal tubules (in a part of the cell called the endoplasmic reticulum). The abnormal protein builds up in the cell and causes it slowly to die, leading to an ultimate loss of the kidney's function manifesting as a disease state.[10][11]
MKD1/Mucin-1 kidney disease
Mucin-1 kidney disease (MKD) is due to a mutation within the MUC1 gene, which is located on chromosome 1.[12] The mucin-1 protein is involved in the creation of a mucus-like substance that coats the surface of different small tubules in the body, it is expressed on distal tubular cells in the kidney. Mucin-1 kidney disease (MKD) is caused by a mutation in the MUC1 gene.[12]
This mutation alters the genetic sequence in the MUC1 gene, resulting in a new, mutated protein, furthermore, The disease is extremely rare and its prevalence is unknown. This disease is autosomal dominant, meaning that it is characterized by a 50% chance of inheritance and slowly progressive chronic kidney disease that leads to the need for dialysis or a kidney transplant. People with a mutation in this gene can have a variable rate of loss of kidney function, with some individuals going on dialysis in their thirties while others may not go on dialysis until later in life.[10][13]
Earlier on the condition was called medullary cystic kidney disease type 1. It was named this because some people with the disease had cysts (small holes) in the middle (medulla) of their kidneys. It has since then been found that these cysts are uncommon and are not found in the majority of the patients with MUC1 mutations. For this reason, this name has been abandoned, scientists (part of the Kidney Dialysis Initiatives and Global Outcomes group) specializing in this disease came together formally and created an official name for this and similar conditions.[6]
This condition was designated as Autosomal Dominant Tubulointerstitial Kidney Disease (ADTKD). In autosomal dominant inheritance only one parent need be affected to pass down the disorder. Tubulointerstitial is used because the problems that occur in this disease result as a consequence of atrophy, of tubules, of the loop of Henle and fibrosis of the medullary interstitium.
MKD2/Uromodulin kidney disease
Medullary cystic kidney disease type 2 is due to mutations in a gene named UMOD on chromosome 16 that encodes a protein called uromodulin[11][14] This disease is also autosomal dominant, meaning that it is characterized by a 50% chance of inheritance and slowly progressive chronic kidney disease that leads to the need for dialysis or a kidney transplant. People with this disease suffer from gout relatively early in life. Mutations in this gene can lead to a variable rate of loss of kidney function.[8]
In MKD2 inheritance, just as MKD1, autosomal dominant indicates that 50% of children of an affected person will inherit the disease. "Tubulointerstitial" is used because the problems that occur in this disease happen in the compartment of the kidney known as the tubulointerstitium, a shorter name of the disease is used by most individuals - uromodulin kidney disease (UKD).[15]
Diagnosis
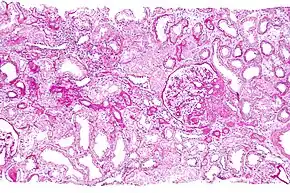
The diagnosis of medullary cystic kidney disease can be done via a physical exam.[4] Further tests/exams are as follows:[1]
- A routine blood test called the serum creatinine can be done. Creatinine is a breakdown product from the muscle, as kidney function declines, the amount of blood creatinine goes up. Thus, most affected individuals have no symptoms of MCKD, but find that they have the condition due to an elevation in the blood creatinine level.[16]
- Affected individuals also have an elevation in the blood uric acid level. In MCKD, the kidney has difficulty getting rid of uric acid. One can find out that the uric acid level in the blood is high when a blood test is done. Gout is caused by high uric acid levels, and thus patients often have gout.[17]
- A kidney ultrasound in this condition usually shows normal or small sized kidneys (occasionally cysts are present). However, since cysts are present in many normal individuals, these cysts are not helpful in making a diagnosis, therefore a kidney biopsy can be done to determine if the individual has this disease. Kidney biopsy is a procedure where a needle is inserted into the kidney and removes a small piece of kidney tissue. This tissue is then examined under a microscope.[18][19]
- Definitive testing and diagnosis of MCKD can be made by analyzing the UMOD gene for mutations, this can be done by a blood test.[20]
Treatment
In terms of treatment/management for medullary cystic kidney disease, at present there are no specific therapies for this disease, and there are no specific diets known to slow progression of the disease. However, management for the symptoms can be dealt with as follows: erythropoietin is used to treat anemia, and growth hormone is used when growth becomes an issue. Additionally, a kidney transplant may be needed at some point. Finally, foods that contain potassium and phosphate must be reduced.[1]
Research
Scientists from the Broad Institute, Cambridge, Massachusetts identified the genetic cause of UKD as mutations in the MUC1 gene.[12]
See also
References
- 1 2 3 4 5 6 "Medullary Cystic Disease Treatment & Management: Medical Care, Surgical Care, Consultations". eMedicine. MedScape. Retrieved 15 June 2016.
- 1 2 "OMIM Entry - # 174000 - MEDULLARY CYSTIC KIDNEY DISEASE 1; MCKD1". omim.org. Retrieved 2 January 2018.
- 1 2 "OMIM Entry - # 603860 - MEDULLARY CYSTIC KIDNEY DISEASE 2; MCKD2". omim.org. Retrieved 2 January 2018.
- 1 2 3 "Medullary cystic kidney disease: MedlinePlus Medical Encyclopedia". www.nlm.nih.gov. Retrieved 2016-06-03.
- ↑ Torres, Vincent E.; Harris, Peter C. (2016). "Cystic Diseases of the Kidney". In Skorecki, Karl; Chertow, Glenn M.; Marsden, Philip A; Taal, Maarten W.; Yu, Alan S.L. (eds.). Brenner and Rector's The Kidney (10th ed.). Philadelphia, PA: Elsevier. p. 1504. ISBN 978-1-4557-4836-5.
Because the development of medullary cysts is neither an early nor a typical feature of the disease, many now consider the term medullary cystic kidney disease to be a misnomer and have proposed the term autosomal dominant tubulointerstitial kidney disease (ADTKD) to refer to a group of diseases caused by at least four genes: MUC1 encoding the mucoprotein mucin-1 (MUC-1) (chromosomal location 1q22); UMOD encoding uromodulin, also known as Tamm-Horsfall protein (chromosome 16p12.3); HNF1B encoding hepatocyte nuclear factor-1β (chromosome 17q12); and REN encoding renin (chromosome 1q32.1).
- 1 2 Eckardt, KU; Alper, SL; Antignac, C; Bleyer, AJ; Chauveau, D; Dahan, K; Deltas, C; Hosking, A; Kmoch, S; Rampoldi, L; Wiesener, M; Wolf, MT; Devuyst, O (October 2015). "Autosomal dominant tubulointerstitial kidney disease: diagnosis, classification, and management--A KDIGO consensus report". Kidney International. 88 (4): 676–83. doi:10.1038/ki.2015.28. PMID 25738250. – via ScienceDirect (Subscription may be required or content may be available in libraries.)
- ↑ Kmoch, Stanislav; Živná, Martina; Bleyer, Anthony (1993). "Autosomal Dominant Tubulointerstitial Kidney Disease, REN-Related". Autosomal Dominant Tubulointerstitial Kidney Disease, REN-Related (ADTKD-REN). GeneReviews. University of Washington, Seattle. Retrieved June 11, 2016.
- 1 2 Bleyer, AJ.; Woodard, AS.; Shihabi, Z.; Sandhu, J.; Zhu, H.; Satko, SG.; Weller, N.; Deterding, E.; McBride, D (Jun 2003). "Clinical characterization of a family with a mutation in the uromodulin (tamm-horsfall glycoprotien) gene". Kidney Int. 64 (1): 36–42. doi:10.1046/j.1523-1755.2003.00081.x. PMID 12787393.
- ↑ "Gout: MedlinePlus". www.nlm.nih.gov. Retrieved 2016-06-15.
- 1 2 Reference, Genetics Home. "medullary cystic kidney disease type 1". Genetics Home Reference. Retrieved 2016-06-15.
- 1 2 Reference, Genetics Home. "uromodulin-associated kidney disease". Genetics Home Reference. Retrieved 2016-06-15.
- 1 2 3 Kirby, A. (Mar 2013). "Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing". Nature Genetics. 45 (3): 299–303. doi:10.1038/ng.2543. PMC 3901305. PMID 23396133.
- ↑ Bleyer, Anthony J.; Kmoch, Stanislav (1993-01-01). "Autosomal Dominant Tubulointerstitial Kidney Disease, MUC1-Related". In Pagon, Roberta A.; Adam, Margaret P.; Ardinger, Holly H.; Wallace, Stephanie E.; Amemiya, Anne; Bean, Lora J.H.; Bird, Thomas D.; Fong, Chin-To; Mefford, Heather C. (eds.). Autosomal Dominant Tubulointerstitial Kidney Disease, MUC1-Related (ADTKD-MUC1). Seattle (WA): University of Washington, Seattle. PMID 23946964.update 2013
- ↑ Hart, T. C.; Gorry, M. C.; Hart, P. S.; Woodard, A. S.; Shihabi, Z.; Sandhu, J.; Shirts, B.; Xu, L.; Zhu, H. (2002-12-01). "Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy". Journal of Medical Genetics. 39 (12): 882–892. doi:10.1136/jmg.39.12.882. ISSN 1468-6244. PMC 1757206. PMID 12471200.
- ↑ Bleyer, Anthony J.; Hart, P. Suzanne (1993-01-01). "Autosomal Dominant Tubulointerstitial Kidney Disease, UMOD-Related". In Pagon, Roberta A.; Adam, Margaret P.; Ardinger, Holly H.; Wallace, Stephanie E.; Amemiya, Anne; Bean, Lora J.H.; Bird, Thomas D.; Fong, Chin-To; Mefford, Heather C. (eds.). Autosomal Dominant Tubulointerstitial Kidney Disease, UMOD-Related (ADTKD-UMOD). Seattle (WA): University of Washington, Seattle. PMID 20301530.update 2013
- ↑ "Creatinine blood test: MedlinePlus Medical Encyclopedia". www.nlm.nih.gov. Retrieved 2016-06-15.
- ↑ "Uric acid - blood : MedlinePlus Medical Encyclopedia". www.nlm.nih.gov. Retrieved 2016-06-15.
- ↑ "Kidney Biopsy". www.niddk.nih.gov. Retrieved 2016-06-15.
- ↑ "Abdominal ultrasound: MedlinePlus Medical Encyclopedia". www.nlm.nih.gov. Retrieved 2016-06-15.
- ↑ Reference, Genetics Home. "UMOD". Genetics Home Reference. Retrieved 2016-06-15.
Further reading
- Hateboer, Nick (2001). "Confirmation of a gene locus for medullary cystic kidney disease (MCKD2) on chromosome 16p12". Kidney International. 60 (4): 1233–1239. doi:10.1046/j.1523-1755.2001.00932.x. PMID 11576337.
- Hildebrandt, Friedhelm; Otto, Edgar (1 September 2000). "Molecular Genetics of Nephronophthisis and Medullary Cystic Kidney Disease". Journal of the American Society of Nephrology. 11 (9): 1753–1761. ISSN 1046-6673. PMID 10966501. Retrieved 15 June 2016.