Polycystic kidney disease
| Polycystic kidney disease | |
|---|---|
| Other names: Polycystic kidney syndrome, kidney - polycystic[1] | |
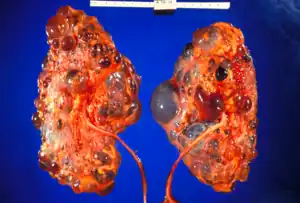 | |
| Severely affected polycystic kidneys removed at time of transplantation | |
| Specialty | Nephrology |
| Symptoms | Flank pain, blood in the urine, headaches, growth failure[2][3] |
| Types | Autosomal dominant polycystic kidney disease (ADPKD), autosomal recessive polycystic kidney disease (ARPKD)[4] |
| Causes | Genetic mutation[4] |
| Risk factors | Family history[4] |
| Diagnostic method | Medical imaging, genetic testing[2][3] |
| Differential diagnosis | Multiple benign simple cysts, medullary sponge kidney, tuberous sclerosis[5] |
| Treatment | Lifestyle changes, blood pressure medication, dialysis, kidney transplant[4] |
| Prognosis | ARPKD 30% risk of early death[3] |
| Frequency | 1 in 700 (ADPKD), 1 in 20,000 (ARPKD)[4] |
Polycystic kidney disease (PKD, PCKD) is a genetic disorder that results in many cysts growing within the kidneys.[4] They may form before birth, during childhood, or in adulthood.[6] When cysts develop before birth growth failure or breathing problems may occur.[3] Otherwise symptoms may include flank pain, blood in the urine, and headaches.[2] Complications may include kidney failure, high blood pressure, liver cysts, aortic aneurysm, and cerebral aneurysm.[4]
It occurs as a result of a genetic mutation.[4] Generally this is inherited from a persons parents though may rarely occur spontaneously during early development.[4] The cysts form from non-functioning tubules that have fluid pumped into them.[7] There are two types autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (ARPKD).[4] The diagnosis maybe confirmed by medical imaging or genetic testing.[2][3]
There is no cure.[2][3] Treatment may include lifestyle changes and medications to reduce blood pressure such as ACE inhibitors.[4] Kidney failure may be managed with dialysis or kidney transplant.[4] About 30% of those with ARPKD die within the first few weeks of life.[3] More than half of people with ADPKD develop kidney failure by the age of 70.[2]
PKD affects about half a million people in the United States.[4] ADPKD affects about 1 in 700 people and ARPKD affects about 1 in 20,000 children.[4] Males and females are affected equally frequently[4] The condition was first described in the 16th and 17th centuries.[8]
Signs and symptoms
Signs and symptoms include high blood pressure, headaches, abdominal pain, blood in the urine, and excessive urination.[1] Other symptoms include pain in the back, and cyst formation (renal and other organs).[9]
Most cases progress to bilateral disease in adulthood.[10]
Cause
PKD is caused by abnormal genes which produce a specific abnormal protein which has an adverse effect on tubule development. PKD is a general term for two types, each having their own pathology and genetic cause: autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (ARPKD).[11][12]
Autosomal dominant
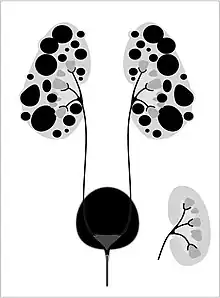
Autosomal dominant polycystic kidney disease (ADPKD) is the most common of all the inherited cystic kidney diseases[10][13][14] with an incidence of 1:500 live births.[10][14] Studies show that 10% of end-stage kidney disease (ESKD) patients being treated with dialysis in Europe and the U.S. were initially diagnosed and treated for ADPKD.[10][12]
Genetic mutations in any of the three genes PKD1, PKD2, and PKD3 have similar phenotypical presentations.
- Gene PKD1 is located on chromosome 16 and codes for a protein involved in regulation of cell cycle and intracellular calcium transport in epithelial cells, and is responsible for 85% of the cases of ADPKD.
- A group of voltage-linked cation channels, with inward selectivity for K>Na>>Ca and outward selectivity for Ca2+ ≈ Ba2+ > Na+ ≈ K+, are coded for by PKD2 on chromosome 4
- PKD3 recently appeared in research papers as a postulated third gene.[10][13] Fewer than 10% of cases of ADPKD appear in non-ADPKD families. Cyst formation begins in utero from any point along the nephron, although fewer than 5% of nephrons are thought to be involved. As the cysts accumulate fluid, they enlarge, separate entirely from the nephron, compress the neighboring kidney parenchyma, and progressively compromise kidney function.[12]
Autosomal recessive
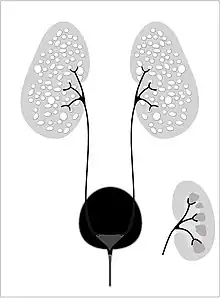
Autosomal recessive polycystic kidney disease (ARPKD) (OMIM #263200) is the lesser common of the two types of PKD, with an incidence of 1:20,000 live births and is typically identified in the first few weeks after birth. Unfortunately, the kidneys are often underdeveloped resulting in a 30% death rate in newborns with ARPKD. PKHD1 is involved.[10][12]
Mechanism
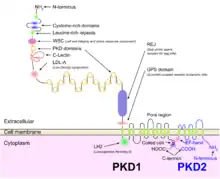
Both autosomal dominant and autosomal recessive polycystic kidney disease cyst formation are tied to abnormal cilia-mediated signaling. The polycystin-1 and polycystin-2 proteins appear to be involved in both autosomal dominant and recessive polycystic kidney disease due to defects in both proteins.[15] Both proteins have communication with calcium channel proteins, and causes reduction in resting (intracellular) calcium and endoplasmic reticulum storage of calcium.[16]
The disease is characterized by a ‘second hit’ phenomenon, in which a mutated dominant allele is inherited from a parent, with cyst formation occurring only after the normal, wild-type gene sustains a subsequent second genetic ‘hit’, resulting in renal tubular cyst formation and disease progression.[17]
PKD results from defects in the primary cilium, an immotile, hair-like cellular organelle present on the surface of most cells in the body, anchored in the cell body by the basal body.[17] In the kidney, primary cilia have been found to be present on most cells of the nephron, projecting from the apical surface of the renal epithelium into the tubule lumen. The cilia were believed to bend in the urine flow, leading to changes in signalling, however this has since been shown to be an experimental error (the bending of cilia was an artifact of focal plane compensation, and also the actual effect on micturition by severe hypertension and cardiac arrest) and that bending of cilia does not contribute to alterations in Ca flux. While it is not known how defects in the primary cilium lead to cyst development, it is thought to possibly be related to disruption of one of the many signaling pathways regulated by the primary cilium, including intracellular calcium, Wnt/β-catenin, cyclic adenosine monophosphate (cAMP), or planar cell polarity (PCP). Function of the primary cilium is impaired, resulting in disruption of a number of intracellular signaling cascades which produce differentiation of cystic epithelium, increased cell division, increased apoptosis, and loss of resorptive capacity.[12][17]
Diagnosis
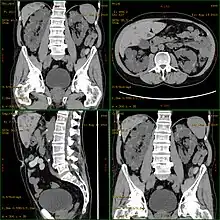
Polycystic kidney disease can be ascertained via a CT scan of abdomen, as well as, an MRI and ultrasound of the same area. A physical exam/test can reveal enlarged liver, heart murmurs and elevated blood pressure[1]
Treatment

If and when the disease progresses enough in a given case, the nephrologist or other practitioner and the patient will have to decide what form of renal replacement therapy will be used to treat end-stage kidney disease (kidney failure, typically stage 4 or 5 of chronic kidney disease).[18]
That will either be some form of dialysis, which can be done at least two different ways at varying frequencies and durations (whether it is done at home or in the clinic depends on the method used and the patient's stability and training) and eventually, if they are eligible because of the nature and severity of their condition and if a suitable match can be found, unilateral or bilateral kidney transplantation.[18]
A Cochrane Review study of autosomal dominant polycystic kidney disease made note of the fact that it is important at all times, while avoiding antibiotic resistance, to control infections of the cysts in the kidneys, and if affected, the liver, when needed for a certain duration to combat infection, by using, "bacteriostatic and bacteriocidal drugs".[12][18]
Prognosis
ADPKD individuals might have a normal life; conversely, ARPKD can cause kidney dysfunction and can lead to kidney failure by the age of 40–60. ADPKD1 and ADPKD2 are very different, in that ADPKD2 is much milder.[19]
Currently, there are no therapies proven effective to prevent the progression of ADPKD.[20]
Epidemiology
PKD is one of the most common hereditary diseases in the United States, affecting more than 600,000 people. It is the cause of nearly 10% of all end-stage renal disease. It equally affects men, women, and all races.[21] PKD occurs in some animals as well as humans.[22][23]
References
- 1 2 3 "Polycystic kidney disease". MedlinePlus Medical Encyclopedia. Archived from the original on 2016-07-05. Retrieved 2015-07-30.
- 1 2 3 4 5 6 "Autosomal Dominant Polycystic Kidney Disease | NIDDK". National Institute of Diabetes and Digestive and Kidney Diseases. Archived from the original on 10 January 2021. Retrieved 24 January 2021.
- 1 2 3 4 5 6 7 "Autosomal Recessive Polycystic Kidney Disease | NIDDK". National Institute of Diabetes and Digestive and Kidney Diseases. Archived from the original on 25 January 2021. Retrieved 24 January 2021.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 "What Is Polycystic Kidney Disease? | NIDDK". National Institute of Diabetes and Digestive and Kidney Diseases. Archived from the original on 21 January 2021. Retrieved 23 January 2021.
- ↑ Akbar, S; Bokhari, SRA (January 2020). "Polycystic Kidney Disease". PMID 30422529.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ Cramer MT, Guay-Woodford LM (2015). "Cystic kidney disease: a primer". Adv Chronic Kidney Dis. 22 (4): 297–305. doi:10.1053/j.ackd.2015.04.001. PMID 26088074.
- ↑ Subramanian, S; Ahmad, T (January 2020). "Polycystic Kidney Disease Of Childhood". PMID 30725822.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ Kenny, Thomas D.; Beales, Philip L. (2013). Ciliopathies: A reference for clinicians. OUP Oxford. p. PT249. ISBN 978-0-19-163337-9. Archived from the original on 2021-08-29. Retrieved 2021-01-24.
- ↑ "Polycystic Kidney Disease". www.niddk.nih.gov. Archived from the original on 2017-01-04. Retrieved 2015-07-31.
- 1 2 3 4 5 6 Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A (2006). "Renal cystic diseases: a review". Advanced Anatomic Pathology. 13 (1): 26–56. doi:10.1097/01.pap.0000201831.77472.d3. PMID 16462154.
- ↑ Porth, Carol (2011-01-01). Essentials of Pathophysiology: Concepts of Altered Health States. Lippincott Williams & Wilkins. ISBN 9781582557243.
- 1 2 3 4 5 6 Phua, YL; Ho, J (April 2015). "MicroRNAs in the pathogenesis of cystic kidney disease". Current Opinion in Pediatrics. 27 (2): 219–26. doi:10.1097/mop.0000000000000168. PMC 4409326. PMID 25490692.
- 1 2 Torres VE; Harris PC; Pirson Y (2007). "Autosomal dominant polycystic kidney disease". Lancet. 369 (9569): 1287–301. doi:10.1016/S0140-6736(07)60601-1. PMID 17434405.
- 1 2 Simons M; Walz G (2006). "Polycystic kidney disease: cell division with a c(l)ue?". Kidney International. 70 (5): 854–864. doi:10.1038/sj.ki.5001534. PMID 16816842.
- ↑ Halvorson, Christian R; Bremmer, Matthew S; Jacobs, Stephen C (2010-06-24). "Polycystic kidney disease: inheritance, pathophysiology, prognosis, and treatment". International Journal of Nephrology and Renovascular Disease. 3: 69–83. doi:10.2147/ijnrd.s6939. ISSN 1178-7058. PMC 3108786. PMID 21694932.
- ↑ Johnson, Richard J.; Feehally, John; Floege, Jurgen (2014-09-05). Comprehensive Clinical Nephrology: Expert Consult - Online. Elsevier Health Sciences. ISBN 9780323242875.
- 1 2 3 Halvorson, C. R.; Bremmer, M. S.; Jacobs, S. C. (2014-05-24). Comprehensive Clinical Nephrology: Polycystic Kidney Disease: Inheritance, pathophysiology, prognosis, and treatment - Online. International Journal of Nephrology and Renovascular Disease. Vol. 3. Int J nephrol Renovasc Dis. pp. 69–83. ISBN 9780323242875. PMC 3108786. PMID 21694932.
- 1 2 3 Montero, Nuria; Sans, Laia; Webster, Angela C; Pascual, Julio (29 January 2014). "Interventions for infected cysts in people with autosomal dominant polycystic kidney disease". Cochrane Database of Systematic Reviews. doi:10.1002/14651858.cd010946.
- ↑ "Polycystic Kidney Disease: Practice Essentials, Background, Pathophysiology". 2018-07-20. Archived from the original on 2015-08-05. Retrieved 2015-07-30.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ Bolignano D, Palmer SC, Ruospo M, Zoccali C, Craig JC, Strippoli GF (2015). "Interventions for preventing the progression of autosomal dominant polycystic kidney disease". Cochrane Database Syst Rev (7): CD010294. doi:10.1002/14651858.CD010294.pub2. PMID 26171904.
- ↑ Tamparo, Carol (2011). Fifth Edition : Diseases of the Human Body. Philadelphia, PA: F.A. Davis Company. p. 443. ISBN 978-0-8036-2505-1.
- ↑ "Polycystic kidney disease (PKD): Gene test and negative register". International Cat Care. Archived from the original on 17 November 2016. Retrieved 2 November 2014.
- ↑ "PKD - Polycystic Kidney Disease - British Shorthair". Antagene. Archived from the original on 17 August 2018. Retrieved 2 November 2014.
Further reading
- Chapin, Hannah C.; Caplan, Michael J. (2010-11-15). "The cell biology of polycystic kidney disease". The Journal of Cell Biology. 191 (4): 701–710. doi:10.1083/jcb.201006173. ISSN 0021-9525. PMC 2983067. PMID 21079243.
- Harris, Peter C.; Torres, Vicente E. (2009-01-01). "Polycystic Kidney Disease". Annual Review of Medicine. 60: 321–337. doi:10.1146/annurev.med.60.101707.125712. ISSN 0066-4219. PMC 2834200. PMID 18947299.
- Press, Dove (2010). "Polycystic kidney disease: inheritance, pathophysiology, prognosis, an | IJNRD". International Journal of Nephrology and Renovascular Disease. 3: 69–83. doi:10.2147/IJNRD.S6939. PMC 3108786. PMID 21694932.
External links
| Classification | |
|---|---|
| External resources |