Alström syndrome
| Alström syndrome | |
|---|---|
| Other names: Alström–Hallgren syndrome | |
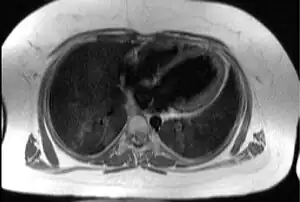 | |
| Transaxial image of the chest at the level of the heart illustrating the typical degree of subcutaneous and epicardial adipose tissue (shown in white) in a patient with Alström syndrome; hence the difficulty in imaging by echocardiography | |
Alström syndrome (AS), also called Alström–Hallgren syndrome,[1] is a very rare autosomal recessive genetic disorder characterised by childhood obesity and multiple organ dysfunction. Symptoms include early-onset type 2 diabetes, cone-rod dystrophy resulting in blindness, sensorineural hearing loss and dilated cardiomyopathy. Endocrine disorders typically also occur, such as hypergonadotrophic hypogonadism and hypothyroidism, as well as acanthosis nigricans resulting from hyperinsulinemia. Developmental delay is seen in almost half of people with Alström syndrome.[2]
It is caused by mutations in the gene ALMS1, which is involved in the formation of cellular cilia, making Alström syndrome a ciliopathy. At least 239 disease-causing mutations in ALMS1 have been described as of 2015.[3] Alström syndrome is sometimes confused with Bardet–Biedl syndrome, another ciliopathy which has similar symptoms, but Bardet–Biedl syndrome tends to have later onset in its symptoms, includes polydactyly and is caused by mutations in BBS genes.[4]
There is no cure for Alström syndrome. Treatments target the individual symptoms and can include diet, corrective lenses, hearing aids, medications for diabetes and heart issues and dialysis and transplantation in the case of kidney or liver failure. Prognosis varies depending on the specific combination of symptoms, but individuals with Alström syndrome rarely live beyond 50.[5]
At least 900 cases have been reported.[6] Prevalence is fewer than 1 in 1,000,000 individuals in the general population,[4] but the disorder is much more common in Acadians, both in Nova Scotia and Louisiana.[7] It was first described by Swedish psychiatrist Carl-Henry Alström and his three associates, B. Hallgren, I. B. Nilsson and H. Asander, in 1959.[8]
Signs and symptoms
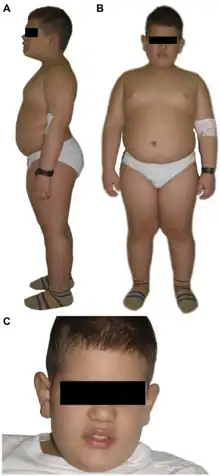
Symptoms for Alström syndrome generally appear during infancy with great variability in age. Some of the symptoms include:[9]
- Heart failure (dilated cardiomyopathy) in over 60% of cases, usually within the first few weeks after birth, but sometimes the onset is in adolescence or adulthood.[10]
- Light sensitivity and vision problems (cone-rod dystrophy) in all cases, usually within 15 months of birth and progressively worsening until about 20 years of age[10]
- Delays in early, developmental milestones in 50% of cases, learning disabilities in about 30% of cases[11]
- Obesity in 100% of cases, apparent by 5 years of age but often apparent in infancy (Alström infants usually have normal birth weights, and by adolescence, weights tend to be in the high-normal to normal range.)
- Nystagmus (usually affects the children), one of the first symptoms to occur which causes involuntary rapid eye movement.
- Mild to moderate bilateral sensorineural hearing loss.
- Type 2 diabetes usually occurs in early childhood.
- Hyperinsulinemia/insulin resistance—development of high level of insulin in blood.
- Hypertriglyceridemia
- Steatosis (fatty liver) and elevated transaminases (liver enzymes) often develop in childhood and can progress in some patients to cirrhosis and liver failure.
- Endocrine dysfunctions may occur where the patient may experience an under or over active thyroid gland, weak growth hormone, increased androgen in females and low testosterone in males.
- Slowly progressive kidney failure can occur in the second to fourth decade of life.
Cause

Alström syndrome is caused by a mutation in the ALMS1 gene, located on the short arm of chromosome 2 (2p13.2). The gene mutation is inherited as an autosomal recessive trait. This means both parents have to pass a defective copy of the ALMS1 gene in order for their child to have the syndrome, even though the parents may not show signs or symptoms of the condition.[12][13]
The ALMS1 gene contains instructions to encode a specific protein known as ALMS1. The protein then is involved in ciliary function, cell cycle control and intracellular transport. In addition, the protein is expressed in all organ tissues of the body. It has a role in the proper function, maintenance and formation of cilia, which are found in all types of cells in the body.[14] At least 239 disease-causing mutations in ALMS1 have been described as of 2015.[3]
Most of these mutations have led to the production of a dysfunctional version of the ALSM1 protein which are present in tissues, but at low levels.[15]
Diagnosis
It is possible to clinically detect Alström syndrome in infancy, but more frequently, it is detected much later, as doctors tend to detect symptoms as separate problems. Currently, Alström syndrome is often diagnosed clinically, since genetic testing is costly and only available on a limited basis.[6]
A physical examination would be needed to properly diagnose the patient. Certain physical characteristics can determine if the patient has some type of genetic disorder. Usually, a geneticist would perform the physical examination by measuring the distance around the head, distance between the eyes and the length of arms and legs. In addition, examinations for the nervous system or the eyes may be performed. Various imaging studies like computerized tomography scans (CT), Magnetic Resonance Imaging (MRI) or X-rays are used to see the structures within the body.[6]
Family and personal medical history are required. Information about the health of an individual is crucial because it provides traces to a genetic diagnosis.
Laboratory tests, particularly genetic testing, are performed to diagnose genetic disorders. Some of the types of genetic testing are molecular, biochemical and chromosomal. Other laboratory tests performed may measure levels of certain substances in urine and blood that can also help suggest a diagnosis.
Related disorders
Recent findings in genetic research have suggested that a large number of genetic disorders, both genetic syndromes and genetic diseases, that were not previously identified in the medical literature as related, may be, in fact, highly related in the genetypical root cause of the widely varying, phenotypically-observed disorders. Thus, Alstrom syndrome is a ciliopathy. Other known ciliopathies include primary ciliary dyskinesia, Bardet–Biedl syndrome, polycystic kidney and liver disease, nephronophthisis, Meckel–Gruber syndrome and some forms of retinal degeneration.[16]
Diagnostic criteria
Marshall JD et al. provided a comprehensive guidance for diagnostic criteria in their 2007 publication.
Birth – 2 years:
Minimum diagnosis requires 2 major criteria or 1 major and 2 minor criteria.
Major criteria are:
- ALMS1 mutation in 1 allele and/or family history of Alström syndrome
- Vision pathology (nystagmus, photophobia).
Minor criteria are:
- Obesity
- Dilated cardiomyopathy with congestive heart failure.
Other variable supportive evidence: Recurrent pulmonary infections, normal digits, delayed developmental milestones.
At 3–14 years of age:
2 major criteria or 1 major and 3 minor criteria.
Major criteria are:
- ALMS1 mutation in 1 allele and/or family history of Alström syndrome,
- Vision pathology (nystagmus, photophobia, diminished acuity). If old enough for testing: cone dystrophy by ERG.
Minor criteria:
- Obesity and/or insulin resistance and/or Type 2 Diabetes
- History of dilated cardiomyopathy with congestive heart failure
- Hearing loss
- Liver dysfunction
- Kidney failure
- Advanced bone age
Variable supportive evidence: Recurrent pulmonary infections, normal digits, delayed developmental milestones, hyperlipidemia, scoliosis, flat wide feet hypothyroidism, hypertension, recurrent urinary tract infection, growth hormone deficiency.
Presentation 15 years – adulthood:
2 major and 2 minor criteria or 1 major and 4 minor criteria.
Major criteria are:
- ALMS1 mutation in 1 allele and/or family history of Alström syndrome.
- Vision pathology (history of nystagmus in infancy/childhood, legal blindness, cone and rod dystrophy by ERG).
Minor criteria:
- Obesity and/or insulin resistance and/or Type 2 Diabetes
- History of dilated cardiomyopathy with congestive heart failure.
- Hearing loss
- Hepatic dysfunction
- Renal failure
- Short stature
- Males: hypogonadism, Females: irregular menses and/or hyperandrogenism
Other supportive features:
Recurrent pulmonary infections, normal digits, history of developmental delay, hyperlipidemia, scoliosis, flat wide feet, hypothyroidism, hypertension, recurrent urinary tract infections/urinary dysfunction, growth hormone deficiency, alopecia.
Prevention
Prevention for Alström syndrome is considered to be harder compared to other diseases/syndromes because it is an inherited condition. However, there are other options that are available for parents with a family history of Alström syndrome. Genetic testing and counseling are available where individuals are able to meet with a genetic counselor to discuss risks of having the children with the disease. The genetic counselor may also help determine whether individuals carry the defective ALSM1 gene before the individuals conceive a child. Some of the tests the genetic counselors perform include chorionic villus sampling (CVS), preimplantation genetic diagnosis (PGD) and amniocentesis. With PGD, the embryos are tested for the ALSM1 gene and only the embryos that are not affected may be chosen for implantation via in vitro fertilization.[17]
Treatment
There is no cure for Alström syndrome; however, there are treatment aims to reduce the symptoms and prevent further complications. Some of these treatment aims include:[13][17][5]
- Corrective lenses: tinted lenses that help with the sensitivity from bright lights. The patients may have to adapt to reading in Braille, use adaptive equipment, mobility aids and adaptive computing skills.
- Education: patients with Alström syndrome suffering from intellectual disabilities must have access to education. They must be able to receive free and appropriate education. Some Alström syndrome patients are educated in normal classrooms. Other patients have to take special education classes or attend to specialized schools that are prepared to teach children with disabilities. Staff members from schools have to consult with patient's parents or caregivers in order to design an education plan based on the child's needs. In addition, the school may document the progress of the child in order to confirm that the child's needs are being met.
- Hearing aids: the battery-operated devices are available in three styles: behind the ear, in the ear and inside the ear canal. Behind the ear aims for mild-to-profound hearing loss. In the ear aims for mild to severe hearing loss. Lastly, the canal device is aimed for mild to moderately severe hearing loss. Patients that have severe hearing loss may benefit from a cochlear implant.
- Diet: an appropriate and healthy diet is necessary for individuals with Alström syndrome because it could potentially decreases chances of obesity or diabetes.
- Occupational therapy: the therapist helps the child learn skills to help him or her perform basic daily tasks like eating, getting dressed and communicating with others.
- Physical Activity: exercising reduces chances of being obese and helping control blood sugar levels.
- Dialysis: helps restore filtering function. With hemodialysis, a patient's blood circulates into an external filter and clean. The filtered blood is then returned into the body. With peritoneal dialysis, fluid containing dextrose is introduced into the abdomen by a tube. The solution then absorbs the wastes into the body and is then removed.
- Transplantation: patients that endure a kidney failure may undergo a kidney transplantation.
- Surgery: if the patient endures severe scoliosis or kyphosis, surgery may be required.
Medication
- Antibiotics: patients with lung problems will be prescribed antibiotics because they are more prone to infections like bronchitis.
- Oral diabetes medications: are taken by mouth to treat diabetes. Can be taken combined into a single pill, which may be more effective and convenient for people with diabetes. It is usually taken once or twice daily before meals. Some of these medications includes:
- Meglitinides (repaglinide and nateglinide): taken to stimulate the cells found in the pancreas to release insulin. These drugs are taken by mouth daily before each meal and could cause a drop in blood sugar.[15]
- Metformin (biguanide): decreases the amount blood sugar being released by the liver and by stimulating the cells within muscles to take up blood sugar. Taken twice daily.
- Thiazolidinediones (rosiglitazone and pioglitazone): taken to help insulin work more efficiently in muscle and fat cells causing the liver to release less glucose. Is associated with heart failure.
- Dipeptidyl peptidase IV (DPP-4) inhibitors (sitagliptin): helps with improving blood sugar levels by decreasing the action of an enzyme breaking down GLP-1 (lowers the blood sugar level).
- Injected diabetes medicine: taken by an injection into the fat below the skin. Sometimes referred as subcutaneous injections. Some of these medications include the following:[15]
- Pramlintide (Symlin): is an Amylin agonist. It acts centrally (via the brain) to reduce food intake and blood sugar. It is most commonly used at mealtimes by people with type 1 and type 2 diabetes.
- Exenatide (Byetta): synthetic form of exendin-4 ( a GLP-1 receptor agonist that increases secretion of insulin, decreases the secretion of glucagon from the pancreas and reduces food intake).
- Cholesterol-lowering medications: is necessary when cholesterol levels are high. HMG-CoA reductase inhibitors, also called "statins," effectively lower levels of low-density lipoprotein, cholesterol and triglycerides. High-dose nicotinic acid (niacin) may also reduce cholesterol levels.[18]
- Heart medications: Angiotensin-converting enzyme (ACE) inhibitors, diuretics, digoxin and beta-blockers may help with the management of cardiomyopathy and heart failure.[18]
Prognosis
A prognosis for Alström syndrome is complicated because it widely varies. Any person that has the syndrome have different set of disorders. Permanent blindness, deafness and type 2 diabetes may occur. Liver and kidney failure can progressively get worse. The life expectancy is usually reduced and the patients rarely live past 50 years old.[13][5][19]
Research
The Jackson Laboratory in Bar Harbor, Maine, USA with the University of Southampton, UK isolated the single gene (ALMS1) responsible for Alström syndrome.[6]
Research was conducted in 2014 on Alström syndrome patients regarding degeneration and plasticity of the optic pathway. The functional and structural changes have been investigated on the optic pathway in Alström syndrome by using magnetic resonance imaging to provide better insight on the underlying pathogenic mechanisms. Eleven patients with the syndrome (mean age of 23 years, 5 females, 6 males) underwent a brain MRI. The protocol also included conventional sequences, resting-state functional MRI and diffusion tensor imaging. Results found that patients with Alström syndrome had occipital regions with decreased white matter volume as well as decreased gray matter volume sparing the occipital poles. The diffused fractional anisotropy decreased and the radial diffusivity increased while mean and axial diffusivities were normal. Lastly, the reduced connectivity in the medial visual network was strikingly sparing the occipital poles. The conclusion of the research was that the protean occipital brain changes in patients with Alström syndrome. They are likely to reflect coexistence of diffuse primary myelin derangement, anterograde trans-synaptic degeneration and complex cortical reorganization that affect the posterior and anterior visual cortex.[20]
References
- ↑ Ewerbeck, H. (2012-12-06). Differential Diagnosis in Pediatrics: A Compendium of Symptoms and Findings. Springer Science & Business Media. p. 315. ISBN 9781461260745. Archived from the original on 2021-04-04. Retrieved 2020-12-04.
- ↑ Joy, T; Cao, H; Black, G; Malik, R; Charlton-Menys, V; Hegele, RA; Durrington, PN (21 December 2007). "Alstrom syndrome (OMIM 203800): a case report and literature review". Orphanet Journal of Rare Diseases. 2: 49. doi:10.1186/1750-1172-2-49. PMC 2266715. PMID 18154657.
- 1 2 Marshall, Jan D.; Muller, Jean; Collin, Gayle B.; Milan, Gabriella; Kingsmore, Stephen F.; Dinwiddie, Darrell; Farrow, Emily G.; Miller, Neil A.; Favaretto, Francesca (July 2015). "Alström Syndrome: Mutation Spectrum of ALMS1". Human Mutation. 36 (7): 660–668. doi:10.1002/humu.22796. ISSN 1098-1004. PMC 4475486. PMID 25846608.
- 1 2 "Alström Syndrome". NORD (National Organization for Rare Disorders). Archived from the original on 2021-03-18. Retrieved 2015-12-07.
- 1 2 3 "Alstrom Syndrome". Healthline. Archived from the original on 5 March 2015. Retrieved 2015-12-06.
- 1 2 3 4 "Alström syndrome". Genetics Home Reference. 2015-11-30. Archived from the original on 2020-09-20. Retrieved 2015-12-06.
- ↑ "OMIM Entry - # 203800 - ALSTROM SYNDROME; ALMS". www.omim.org. Archived from the original on 2021-03-22. Retrieved 2019-07-16.
- ↑ Alstrom, C. H.; Hallgren, B.; Nilsson, L. B.; Asander, H. (1959). "Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: a specific syndrome (not hitherto described) distinct from the Laurence-Moon-Bardet-Biedl syndrome: a clinical, endocrinological and genetic examination based on a large pedigree". Acta Psychiatrica et Neurologica Scandinavica. Supplementum. 129: 1–35. ISSN 0365-5067. PMID 13649370.
- ↑ "Alstrom Syndrome". Healthline. Archived from the original on 2015-03-05. Retrieved 2015-12-06.
- 1 2 "Alström Syndrome". Archived from the original on 2021-03-16. Retrieved 2020-12-04.
- ↑ Marshall J, Paisey RB, Carey C, Macdermott S. Alström syndrome. GeneReviews. May 31, 2012; http://www.ncbi.nlm.nih.gov/books/NBK1267/ Archived 2017-01-18 at the Wayback Machine
- ↑ "Alstrom Syndrome". Healthline. Archived from the original on 2015-03-05. Retrieved 2015-12-06.
- 1 2 3 "Alström Syndrome — Symptoms, Diagnosis, Treatment of Alström Syndrome". NY Times Health Information. Archived from the original on 7 March 2016. Retrieved 2015-12-06.
- ↑ "Alström Syndrome". NORD (National Organization for Rare Disorders). Archived from the original on 2021-03-18. Retrieved 2015-11-05.
- 1 2 3 4 "Alstrom syndrome". New Bridge Organic Market Jacksonville. Archived from the original on 10 December 2015. Retrieved 2015-12-06.
- ↑ Badano, Jose L.; Norimasa Mitsuma; Phil L. Beales; Nicholas Katsanis (September 2006). "The Ciliopathies : An Emerging Class of Human Genetic Disorders". Annual Review of Genomics and Human Genetics. 7: 125–148. doi:10.1146/annurev.genom.7.080505.115610. PMID 16722803.
- 1 2 3 "Alstrom syndrome". Nature's Corner. Retrieved 2015-12-06.
- 1 2 "Alstrom syndrome - Clark's Nutrition". www.clarksnutrition.com. Archived from the original on 2015-12-10. Retrieved 2015-11-04.
- ↑ "About Alström Syndrome". www.deafblindinternational.org. Archived from the original on 16 November 2017. Retrieved 2015-12-06.
- ↑ Manara, R.; Citton, V.; Maffei, P.; Marshall, J. D.; Naggert, J. K.; Milan, G.; Vettor, R.; Baglione, A.; Vitale, A. (2015-01-01). "Degeneration and plasticity of the optic pathway in Alström syndrome". AJNR. American Journal of Neuroradiology. 36 (1): 160–5. doi:10.3174/ajnr.A4115. PMID 25355816.
Further reading
- Marshall JD, Beck S, Maffei P, Naggert JK (2007). "Alström syndrome". Eur. J. Hum. Genet. 15 (12): 1193–202. doi:10.1038/sj.ejhg.5201933. PMID 17940554.
External links
| Classification | |
|---|---|
| External resources |
|