Autosomal recessive polycystic kidney disease
| Autosomal recessive polycystic kidney disease | |
|---|---|
| Other names: ARPKD | |
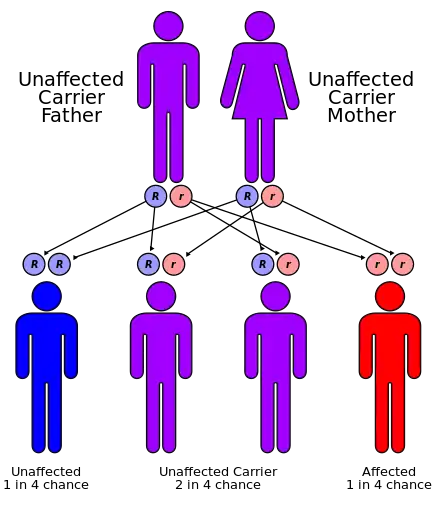 | |
| ARPKD is inherited in an autosomal recessive pattern | |
| Symptoms | Polyuria[1] |
| Causes | Mutations in the PKHD1 gene[2] |
| Diagnostic method | Ultrasound[3] |
| Treatment | Medications for hypertension[4] |
Autosomal recessive polycystic kidney disease (ARPKD) is the recessive form of polycystic kidney disease. It is associated with a group of congenital fibrocystic syndromes.[5] Mutations in the PKHD1 (chromosomal locus 6p12.2) cause ARPKD.[6][7]
Signs and symptoms
Symptoms and signs include abdominal discomfort, polyuria, polydipsia, incidental discovery of hypertension, abdominal mass.[1] The classic presentation for ARPKD is systemic hypertension with progression to end-stage kidney disease (ESKD) by the age of 15. In a typical presentation, a small number of ARPKD sufferers live to adulthood with some kidney function; but with significant deterioration in liver function.[8] This outcome is postulated to result from expression of the polycystic kidney and hepatic disease gene PKHD1, which is located on chromosome 6p.[9] In severe cases, a fetus will present with oligohydramnios and as a result, may present with Potter sequence.
Genetics
The cause of ARPKD is linked to mutations in the PKHD1 gene.[2]

ARPKD is a significant hereditary renal disease in that appears in childhood.[10] The prevalence is estimated to be of 1 in 20,000 live births.[10] With a reported carrier frequency of up to 1:70. The single gene mutation called ‘’PKHD1’’ is fully responsible for the disease presentation of ARPKD.[10] This PKHD1 is located on the human chromosome region 6p21.1-6p12.2.[10] It is also one of the largest genes in the genome as it occupies approximately 450 kb of DNA, and contains at least 86 exons.[10]
It is capable of producing multiple alternatively spliced transcripts.[10] The largest known transcript encodes fibrocystin /polyductin (FPC), which is a large receptor-like integral membrane protein of 4074 amino acids.[10] The structure of the FPC consist of a single transmembrane, a large N-terminal extracellular region, and a short intracellular cytoplasmic domain.[10] The FPC protein is found on the primary cilia of epithelia cells of cortical and medullary collecting ducts and cholangiocytes of bile ducts, and show similarity to polycystins and several other ciliopathy proteins.[10] FPC is also found to be expressed on the basal body and plasma membrane.[10] It is presumed that the large extracellular domain of FPC binds to a ligand(s) that is yet unknown and that is also involved in cell-cell and cell-matrix interactions.[10]
It is known that FPC interacts with ADPKD protein PC2 and may also participate in this regulation pathway of the mechanosensory function of the primary cilia, calcium signaling, and PCP.[10] This is suggesting a common mechanism underlying cystogenesis between ADPKD and ARPKD.[10] The FPC protein is also found on the centrosomes and mitotic spindle and may regulate centrosome duplication and mitotic spindle assembly during cell division.[10] There have been a large number of various single-gene mutations found throughout PKHD1 and are unique to individual families. Most of the patients are compound heterozygotes for PKHD1 mutations.[10] Patients with two nonsense mutations appear to have an earlier onset of the disease.[10]
Diagnosis
Ultrasonography is the primary method to evaluate autosomal recessive polycystic kidney disease, particularly in the perinatal and neonatal stages.[3]
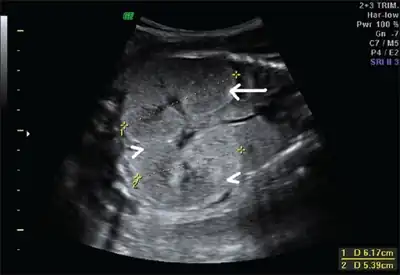 Fetus with bilateral enlarged/hyperechogenic kidneys arrow & diminished cortico-medullary differentiation arrowhead
Fetus with bilateral enlarged/hyperechogenic kidneys arrow & diminished cortico-medullary differentiation arrowhead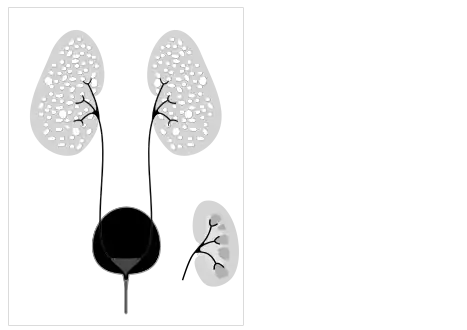 Illustration-Autosomal recessive polycystic kidney disease with a normal kidney inset
Illustration-Autosomal recessive polycystic kidney disease with a normal kidney inset
Differential diagnosis
The differential diagnoses of this condition include:[5]
- Glomerulocystic kidney disease
- Autosomal dominant polycystic kidney disease
- Diffuse cystic dysplasia
Treatment
The treatment options for autosomal recessive polycystic kidney disease, given there is no current cure, are:[4]
- Medications for hypertension
- Medications and/or surgery for pain
- Antibiotics for infection
- Dialysis (if kidney failure is present)
- Kidney transplantation(in serious cases)
References
- 1 2 "Autosomal recessive polycystic kidney disease - Symptoms - NHS Choices". www.nhs.uk. Archived from the original on 2015-07-20. Retrieved 2015-07-28.
- 1 2 "Polycystic kidney disease". Genetics Home Reference. Archived from the original on 2015-07-27. Retrieved 2015-07-28.
- 1 2 "Imaging in Autosomal Recessive Polycystic Kidney Disease: Overview, Radiography, Computed Tomography". 2017-03-30. Archived from the original on 2019-04-14. Retrieved 2021-08-20.
{{cite journal}}: Cite journal requires|journal=(help) - 1 2 "Polycystic Kidney Disease". www.niddk.nih.gov. Archived from the original on 2017-01-04. Retrieved 2015-07-28.
- 1 2 Sweeney, William (1993). "Polycystic Kidney Disease, Autosomal Recessive". Polycystic kidney Disease. NIH. Gene Review. Archived from the original on 16 January 2014. Retrieved 28 July 2015.
- ↑ Bergmann C, Küpper F, Dornia C, Schneider F, Senderek J, Zerres K (March 2005). "Algorithm for efficient PKHD1 mutation screening in autosomal recessive polycystic kidney disease (ARPKD)". Hum. Mutat. 25 (3): 225–31. doi:10.1002/humu.20145. PMID 15706593. S2CID 21321253.
- ↑ Zhang MZ, Mai W, Li C, et al. (February 2004). "PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells". Proc. Natl. Acad. Sci. U.S.A. 101 (8): 2311–6. Bibcode:2004PNAS..101.2311Z. doi:10.1073/pnas.0400073101. PMC 356947. PMID 14983006.
- ↑ Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A (January 2006). "Renal cystic diseases: a review". Adv Anat Pathol. 13 (1): 26–56. doi:10.1097/01.pap.0000201831.77472.d3. PMID 16462154. S2CID 12417947.
- ↑ Sweeney, WE; Avner ED (2006). "Molecular and cellular pathophysiology of autosomal recessive polycystic kidney disease (ARPDK)". Cell and Tissue Research. 326 (3): 671–685. doi:10.1007/s00441-006-0226-0. PMID 16767405. S2CID 33829528.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 Shanahan, James F.; Davis, Kim J. (2015). Harrison's Principles of Internal Medicine (19th ed.). United States of America: McGraw-Hill Education. ISBN 978-0-07-1802161.
Further reading
- Lonergan, Gael J.; Rice, Roy R.; Suarez, Eric S. (2000-05-01). "Autosomal Recessive Polycystic Kidney Disease: Radiologic-Pathologic Correlation". RadioGraphics. 20 (3): 837–855. doi:10.1148/radiographics.20.3.g00ma20837. ISSN 0271-5333. PMID 10835131.
- Rajanna, Dayananda Kumar; Reddy, Anjani; Srinivas, Naren Satya; Aneja, Ankur (2013-03-29). "Autosomal Recessive Polycystic Kidney Disease: Antenatal Diagnosis and Histopathological Correlation". Journal of Clinical Imaging Science. 3: 13. doi:10.4103/2156-7514.109733. ISSN 2156-7514. PMC 3690676. PMID 23814685.
External links
| Classification | |
|---|---|
| External resources |
|