3-Hydroxy-3-methylglutaryl-CoA lyase deficiency
| 3-Hydroxy-3-methylglutaryl-CoA lyase deficiency | |
|---|---|
| Other names: HMG-CoA lyase deficiency or Hydroxymethylglutaric aciduria | |
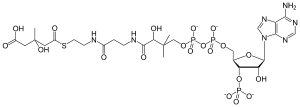 | |
| HMG-CoA | |
3-Hydroxy-3-methylglutaryl-CoA lyase deficiency is an uncommon inherited disorder in which the body cannot properly process the amino acid leucine.[1] Additionally, the disorder prevents the body from making ketones, which are used for energy during fasting.
Signs and symptoms
This disorder usually appears within the first year of life. The signs and symptoms of HMG-CoA lyase deficiency include vomiting, dehydration, lethargy, convulsions, and coma. When episodes occur in an infant or child, blood sugar becomes extremely low (hypoglycemia), and harmful compounds can build up and cause the blood to become too acidic (metabolic acidosis). These episodes are often triggered by an infection, fasting, strenuous exercise, or sometimes other types of stress.
Cause
Mutations in the HMGCL gene cause 3-hydroxy-3-methylglutaryl-CoA lyase deficiency. The enzyme made by the HMGCL gene plays an essential role in breaking down dietary proteins and fats for energy. Specifically, the enzyme is responsible for processing leucine, an amino acid that is part of many proteins. This enzyme also produces ketones during the breakdown of fats. If a mutation in the HMGCL gene reduces or eliminates the activity of this enzyme, the body is unable to process leucine or make ketones properly. A lack of ketones leads to hypoglycemia, and compounds called organic acids (which are formed as products of amino acid and fat breakdown) can cause the blood to become too acidic. Metabolic acidosis and hypoglycemia impair tissue function, especially in the central nervous system.
Diagnosis
The diagnosis of this condition is ascertained via the following:[2]
- Medical exam
- Medical history
- Lab test
- Genetic test
Differential diagnosis
This condition is sometimes mistaken for Reye syndrome, a severe disorder that develops in children while they appear to be recovering from viral infections such as chicken pox or flu. Most cases of Reye syndrome are associated with the use of aspirin during these viral infections.
Treatment
The management of this condition is based on the administration of intravenous 10% glucose and supportive treatment (during metabolic crisis)[3]
Epidemiology
Less than 20 patients with MGA type I have been reported in the literature (Mol Genet Metab. 2011 Nov;104(3):410-3. Epub 2011 Jul 26.)
See also
- 3-hydroxy-3-methylglutaryl-CoA lyase
References
- ↑ Reference, Genetics Home. "HMG-CoA lyase deficiency". Genetics Home Reference. Archived from the original on 2017-02-28. Retrieved 2017-02-27.
- ↑ "HMG CoA lyase deficiency | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 21 October 2021. Retrieved 14 January 2022.
- ↑ RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: 3 hydroxy 3 methylglutaric aciduria". www.orpha.net. Archived from the original on 27 November 2020. Retrieved 14 January 2022.
This article incorporates public domain text from The U.S. National Library of Medicine Archived 2019-02-04 at the Wayback Machine
External links
| Classification |
|
|---|