Sodium phenylbutyrate
Sodium phenylbutyrate, sold under the brand name Buphenyl among others, is a salt of an aromatic fatty acid, 4-phenylbutyrate (4-PBA) or 4-phenylbutyric acid.[5] The compound is used to treat urea cycle disorders, because its metabolites offer an alternative pathway to the urea cycle to allow excretion of excess nitrogen.[6][7]
 | |
| Clinical data | |
|---|---|
| Trade names | Buphenyl, Pheburane, Ammonaps, others |
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| License data |
|
| Pregnancy category |
|
| Routes of administration | By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Metabolism | Liver and kidney to phenylacetic acid |
| Elimination half-life | 0.8 hours (phenylbutyrate), 1.15-1.29 hours (phenylacetate) |
| Excretion | Urine (80-100% as phenylacetylglutamine) |
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.130.318 |
| Chemical and physical data | |
| Formula | C10H11NaO2 |
| Molar mass | 186.186 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
| | |
Sodium phenylbutyrate is also a histone deacetylase inhibitor and chemical chaperone, leading respectively to research into its use as an anti-cancer agent and in protein misfolding diseases such as cystic fibrosis.[5]
Structure and properties
Sodium phenylbutyrate is a sodium salt of an aromatic fatty acid, made up of an aromatic ring and butyric acid. The chemical name for sodium phenylbutyrate is 4-phenylbutyric acid, sodium salt. It forms water-soluble off-white crystals.[2]
Uses
Medical uses
Sodium phenylbutyrate is taken orally or by nasogastric intubation as a tablet or powder, and tastes very salty and bitter. It treats urea cycle disorders, genetic diseases in which nitrogen waste builds up in the blood plasma as ammonia glutamine (a state called hyperammonemia) due to deficiences in the enzymes carbamoyl phosphate synthetase I, ornithine transcarbamylase, or argininosuccinic acid synthetase.[6][2] Uncontrolled, this causes intellectual impairment and early death.[2] Sodium phenylbutyrate metabolites allows the kidneys to excrete excess nitrogen in place of urea, and coupled with dialysis, amino acid supplements and a protein-restricted diet, children born with urea cycle disorders can usually survive beyond 12 months.[2] Patients may need treatment for all their life.[2] The treatment was introduced by researchers in the 1990s, and approved by the U.S. Food and Drugs Administration (FDA) in April 1996.[8][9]
Adverse effects
Nearly 1⁄4 of women may experience an adverse effect of amenorrhea or menstrual dysfunction.[2] Appetite loss is seen is 4% of patients. Body odor due to metabolization of phenylbutyrate affects 3% of patients, and 3% experience unpleasant tastes. Gastrointestinal symptoms and mostly mild indications of neurotoxicity are also seen in less than 2% of patients, among several other reported adverse effects.[2] Administration during pregnancy is not recommended because sodium phenylbutyrate treatment could mimic maternal phenylketonuria due to the production of phenylalanine, potentially causing fetal brain damage.[6]
Urea cycle disorders
Sodium phenylbutyrate administration was discovered to lead to an alternative nitrogen disposal pathway by Dr. Saul Brusilow, Mark Batshaw and colleagues at the Johns Hopkins School of Medicine in the early 1980s, due to some serendipitous discoveries. They had studied ketoacid therapy for another inborn error of metabolism, citrullinemia, in the late 1970s and they noticed that arginine treatment led to an increase of nitrogen in the urine and a drop in ammonia in the blood. The researchers spoke to Norman Radin about this finding, and he remembered a 1914 article on using sodium benzoate to reduce urea excretion. Another 1919 article had used sodium phenylacetate, and so the researchers treated 5 patients with hyperammonemia with benzoate and phenylacetate and published a report in Science.[6][10] In 1982 and 1984, the researchers published on using benzoate and arginine for urea cycle disorders in the NEJM.[6][11][12] Use of sodium phenylbutyrate was introduced in the early 1990s, as it lacks the odor of phenylacetate.[6][13][14]
Chemical chaperone
In cystic fibrosis, a point mutation in the Cystic Fibrosis Transmembrane Conductance Regulator protein, ΔF508-CFTR, causes it to be unstable and misfold, hence trapped in the endoplasmic reticulum and unable to reach the cell membrane. This lack of CFTR in the cell membrane leads to disrupted chloride transport and the symptoms of cystic fibrosis. Sodium phenylbutyrate can act as a chemical chaperone, stabilising the mutant CFTR in the endoplasmic reticulum and allowing it to reach the cell surface.[15]
Histone deacetylase inhibitor
Deriving from its activity as a histone deacetylase inhibitor, sodium phenylbutyrate is under investigation for use as a potential differentiation-inducing agent in malignant glioma and acute myeloid leukaemia,[5] and also for the treatment of some sickle-cell disorders as an alternative to hydroxycarbamide due it inducing expression of fetal hemoglobin to replace missing adult hemoglobin.[16][17] While small-scale investigation is proceeding, there is to date no published data to support the use of the compound in the clinical treatment of cancer, and it remains under limited investigation. Sodium phenylbutyrate is also being studied as a therapeutic option for the treatment of Huntington's disease.[18]
Other
Phenylbutyrate has been associated with longer lifespans in Drosophila.[19]
University of Colorado researchers Dr. Curt Freed and Wenbo Zhou demonstrated that phenylbutyrate stops the progression of Parkinson's disease in mice by turning on a gene called DJ-1 that can protect dopaminergic neurons in the midbrain from dying. As of July 2011 they plan on testing phenylbutyrate for the treatment of Parkinson's disease in humans.[20]
Pharmacology
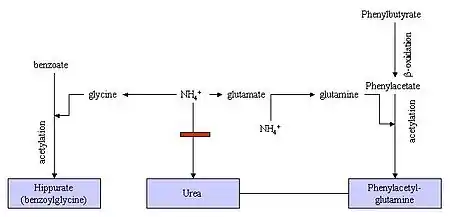
Phenylbutyrate is a prodrug. In the human body it is first converted to phenylbutyryl-CoA and then metabolized by mitochondrial beta-oxidation, mainly in the liver and kidneys, to the active form, phenylacetate.[21] Phenylacetate conjugates with glutamine to phenylacetylglutamine, which is eliminated with the urine. It contains the same amount of nitrogen as urea, which makes it an alternative to urea for excreting nitrogen.[2]
A 5g tablet or powder of sodium phenylbutyrate taken by mouth can be detected in the blood within 15 minutes, and reaches peak concentration in the bloodstream within an hour. It is metabolized into phenylacetate within half an hour.[2]
References
- "Prescription medicines: registration of new chemical entities in Australia, 2017". Therapeutic Goods Administration (TGA). 21 June 2022. Retrieved 9 April 2023.
- "Buphenyl- sodium phenylbutyrate tablet Buphenyl- sodium phenylbutyrate powder". DailyMed. 11 March 2020. Retrieved 19 October 2020.
- "Olpruva - sodium phenylbutyrate kit". DailyMed. 22 December 2022. Retrieved 21 January 2023.
- "Ammonaps EPAR". European Medicines Agency. 17 September 2018. Retrieved 3 January 2023.
- Iannitti T, Palmieri B (September 2011). "Clinical and experimental applications of sodium phenylbutyrate". Drugs in R&D. 11 (3): 227–249. doi:10.2165/11591280-000000000-00000. PMC 3586072. PMID 21902286.
- Batshaw ML, MacArthur RB, Tuchman M (January 2001). "Alternative pathway therapy for urea cycle disorders: twenty years later". The Journal of Pediatrics. 138 (1 Suppl): S46–S54, discussion S54–S55. doi:10.1067/mpd.2001.111836. PMID 11148549.
- Walker V (September 2009). "Ammonia toxicity and its prevention in inherited defects of the urea cycle". Diabetes, Obesity & Metabolism. 11 (9): 823–835. doi:10.1111/j.1463-1326.2009.01054.x. PMID 19531057. S2CID 25998574.
- "Buphenyl: FDA-Approved Drugs". U.S. Food and Drugs Administration (FDA). Retrieved 26 October 2013.
- Buphenyl FDA approval package (PDF). U.S. Food and Drugs Administration (FDA) (Report).
- Brusilow S, Tinker J, Batshaw ML (February 1980). "Amino acid acylation: a mechanism of nitrogen excretion in inborn errors of urea synthesis". Science. 207 (4431): 659–661. Bibcode:1980Sci...207..659B. doi:10.1126/science.6243418. PMID 6243418.
- Batshaw ML, Brusilow S, Waber L, Blom W, Brubakk AM, Burton BK, et al. (June 1982). "Treatment of inborn errors of urea synthesis: activation of alternative pathways of waste nitrogen synthesis and excretion". The New England Journal of Medicine. 306 (23): 1387–1392. doi:10.1056/nejm198206103062303. PMID 7078580.
- Brusilow SW, Danney M, Waber LJ, Batshaw M, Burton B, Levitsky L, et al. (June 1984). "Treatment of episodic hyperammonemia in children with inborn errors of urea synthesis". The New England Journal of Medicine. 310 (25): 1630–1634. doi:10.1056/nejm198406213102503. PMID 6427608.
- Brusilow SW (February 1991). "Phenylacetylglutamine may replace urea as a vehicle for waste nitrogen excretion". Pediatric Research. 29 (2): 147–150. doi:10.1203/00006450-199102000-00009. PMID 2014149.
- Tuchman M, Knopman DS, Shih VE (October 1990). "Episodic hyperammonemia in adult siblings with hyperornithinemia, hyperammonemia, and homocitrullinuria syndrome". Archives of Neurology. 47 (10): 1134–1137. doi:10.1001/archneur.1990.00530100104022. PMID 2222247.
- Chanoux RA, Rubenstein RC (17 July 2012). "Molecular Chaperones as Targets to Circumvent the CFTR Defect in Cystic Fibrosis". Frontiers in Pharmacology. 3 (137): 137. doi:10.3389/fphar.2012.00137. PMC 3398409. PMID 22822398.
- Dover GJ, Brusilow S, Charache S (July 1994). "Induction of fetal hemoglobin production in subjects with sickle cell anemia by oral sodium phenylbutyrate". Blood. 84 (1): 339–343. doi:10.1182/blood.V84.1.339.339. PMID 7517215.
- Trompeter S, Roberts I (February 2009). "Haemoglobin F modulation in childhood sickle cell disease". British Journal of Haematology. 144 (3): 308–316. doi:10.1111/j.1365-2141.2008.07482.x. PMID 19036119. S2CID 23928373.
- Moumné L, Betuing S, Caboche J (October 2013). "Multiple Aspects of Gene Dysregulation in Huntington's Disease". Frontiers in Neurology. 4: 127. doi:10.3389/fneur.2013.00127. PMC 3806340. PMID 24167500.
- Kang HL, Benzer S, Min KT (January 2002). "Life extension in Drosophila by feeding a drug". Proceedings of the National Academy of Sciences of the United States of America. 99 (2): 838–843. Bibcode:2002PNAS...99..838K. doi:10.1073/pnas.022631999. PMC 117392. PMID 11792861.
- Iannitti T, Palmieri B (September 2011). "Clinical and experimental applications of sodium phenylbutyrate". Drugs in R&D. 11 (3): 227–249. doi:10.2165/11591280-000000000-00000. PMC 3586072. PMID 21902286.
The same authors investigated the effects of phenylbutyrate on the accumulation of Parkin-associated endothelin receptor-like receptor (Pael-R), pathologically relevant to the loss of dopaminergic neurons in autosomal recessive juvenile parkinsonism, showing that (i) phenylbutyrate restores the normal expression of Pael-R protein and suppresses ER stress induced by the overexpression of Pael-R; (ii) phenylbutyrate attenuates the activation of ER stress-induced signal transduction pathways and subsequent neuronal cell death; and (iii) phenylbutyrate restores the viability of yeasts that fail to induce an ER stress response under ER stress conditions. These findings lead the author to conclude that phenylbutyrate suppresses ER stress by directly reducing the amount of misfolded protein, including Pael-R accumulated in the ER.[175]
- Kormanik K, Kang H, Cuebas D, Vockley J, Mohsen AW (December 2012). "Evidence for involvement of medium chain acyl-CoA dehydrogenase in the metabolism of phenylbutyrate". Molecular Genetics and Metabolism. 107 (4): 684–689. doi:10.1016/j.ymgme.2012.10.009. PMC 3504130. PMID 23141465.
External links
- "Sodium phenylbutyrate". Drug Information Portal. U.S. National Library of Medicine.