Histone acetylation and deacetylation
Histone acetylation and deacetylation are the processes by which the lysine residues within the N-terminal tail protruding from the histone core of the nucleosome are acetylated and deacetylated as part of gene regulation.
Histone acetylation and deacetylation are essential parts of gene regulation. These reactions are typically catalysed by enzymes with "histone acetyltransferase" (HAT) or "histone deacetylase" (HDAC) activity. Acetylation is the process where an acetyl functional group is transferred from one molecule (in this case, acetyl coenzyme A) to another. Deacetylation is simply the reverse reaction where an acetyl group is removed from a molecule.
Acetylated histones, octameric proteins that organize chromatin into nucleosomes, the basic structural unit of the chromosomes and ultimately higher order structures, represent a type of epigenetic marker within chromatin. Acetylation removes the positive charge on the histones, thereby decreasing the interaction of the N termini of histones with the negatively charged phosphate groups of DNA. As a consequence, the condensed chromatin is transformed into a more relaxed structure that is associated with greater levels of gene transcription. This relaxation can be reversed by deacetylation catalyzed by HDAC activity. Relaxed, transcriptionally active DNA is referred to as euchromatin. More condensed (tightly packed) DNA is referred to as heterochromatin. Condensation can be brought about by processes including deacetylation and methylation.[1]
Mechanism of action
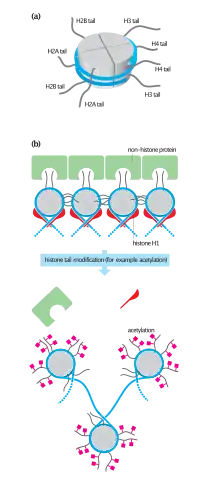
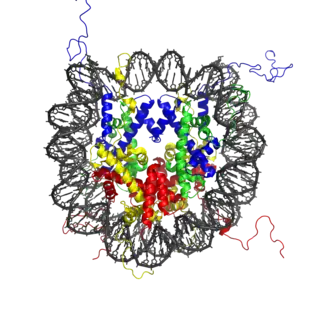
Nucleosomes are portions of double-stranded DNA (dsDNA) that are wrapped around protein complexes called histone cores. These histone cores are composed of 8 subunits, two each of H2A, H2B, H3 and H4 histones. This protein complex forms a cylindrical shape that dsDNA wraps around with approximately 147 base pairs. Nucleosomes are formed as a beginning step for DNA compaction that also contributes to structural support as well as serves functional roles.[2] These functional roles are contributed by the tails of the histone subunits. The histone tails insert themselves in the minor grooves of the DNA and extend through the double helix,[1] which leaves them open for modifications involved in transcriptional activation.[3] Acetylation has been closely associated with increases in transcriptional activation while deacetylation has been linked with transcriptional deactivation. These reactions occur post-translation and are reversible.[3]
The mechanism for acetylation and deacetylation takes place on the NH3+ groups of lysine amino acid residues. These residues are located on the tails of histones that make up the nucleosome of packaged dsDNA. The process is aided by factors known as histone acetyltransferases (HATs). HAT molecules facilitate the transfer of an acetyl group from a molecule of acetyl-coenzyme A (Acetyl-CoA) to the NH3+ group on lysine. When a lysine is to be deacetylated, factors known as histone deacetylases (HDACs) catalyze the removal of the acetyl group with a molecule of H2O.[3][4]
Acetylation has the effect of changing the overall charge of the histone tail from positive to neutral. Nucleosome formation is dependent on the positive charges of the H4 histones and the negative charge on the surface of H2A histone fold domains. Acetylation of the histone tails disrupts this association, leading to weaker binding of the nucleosomal components.[1] By doing this, the DNA is more accessible and leads to more transcription factors being able to reach the DNA. Thus, acetylation of histones is known to increase the expression of genes through transcription activation. Deacetylation performed by HDAC molecules has the opposite effect. By deacetylating the histone tails, the DNA becomes more tightly wrapped around the histone cores, making it harder for transcription factors to bind to the DNA. This leads to decreased levels of gene expression and is known as gene silencing.[5][6][7]
Acetylated histones, the octomeric protein cores of nucleosomes, represent a type of epigenetic marker within chromatin. Studies have shown that one modification has the tendency to influence whether another modification will take place. Modifications of histones can not only cause secondary structural changes at their specific points, but can cause many structural changes in distant locations which inevitably affects function.[8] As the chromosome is replicated, the modifications that exist on the parental chromosomes are handed down to daughter chromosomes. The modifications, as part of their function, can recruit enzymes for their particular function and can contribute to the continuation of modifications and their effects after replication has taken place.[1] It has been shown that, even past one replication, expression of genes may still be affected many cell generations later. A study showed that, upon inhibition of HDAC enzymes by Trichostatin A, genes inserted next to centric heterochromatin showed increased expression. Many cell generations later, in the absence of the inhibitor, the increased gene expression was still expressed, showing modifications can be carried through many replication processes such as mitosis and meiosis.[8]
Histone acetylation/deacetylation enzymes

Histone acetyltransferase (HATs)
Histone Acetyltransferases, also known as HATs, are a family of enzymes that acetylate the histone tails of the nucleosome. This, and other modifications, are expressed based on the varying states of the cellular environment.[2] Many proteins with acetylating abilities have been documented and, after a time, were categorized based on sequence similarities between them. These similarities are high among members of a family, but members from different families show very little resemblance.[9] Some of the major families identified so far are as follows.
GNAT family
General Control Non-Derepressible 5 (Gcn5) –related N-Acetyltransferases (GNATs) is one of the many studied families with acetylation abilities.[10] This superfamily includes the factors Gcn5 which is included in the SAGA, SLIK, STAGA, ADA, and A2 complexes, Gcn5L, p300/CREB-binding protein associated factor (PCAF), Elp3, HPA2 and HAT1.[10][11] Major features of the GNAT family include HAT domains approximately 160 residues in length and a conserved bromodomain that has been found to be an acetyl-lysine targeting motif.[9] Gcn5 has been shown to acetylate substrates when it is part of a complex.[11] Recombinant Gcn5 has been found to be involved in the acetylation of the H3 histones of the nucleosome.[2][11] To a lesser extent, it has been found to also acetylate H2B and H4 histones when involved with other complexes.[2][3][11] PCAF has the ability to act as a HAT protein and acetylate histones, it can acetylate non-histone proteins related to transcription, as well as act as a coactivator in many processes including myogenesis, nuclear-receptor-mediated activation and growth-factor-signaled activation. Elp3 has the ability to acetylate all histone subunits and also shows involvement in the RNA polymerase II holoenzyme.[2]
MYST family
MOZ (Monocytic Leukemia Zinc Finger Protein), Ybf2/Sas3, Sas2 and Tip60 (Tat Interacting Protein) all make up MYST, another well known family that exhibits acetylating capabilities. This family includes Sas3, essential SAS-related acetyltransferase (Esa1), Sas2, Tip60, MOF, MOZ, MORF, and HBO1. The members of this family have multiple functions, not only with activating and silencing genes, but also affect development and have implications in human diseases.[11] Sas2 and Sas3 are involved in transcription silencing, MOZ and TIF2 are involved with the formation of leukemic transclocation products while MOF is involved in dosage compensation in Drosophila. MOF also influences spermatogenesis in mice as it is involved in the expansion of H2AX phosphorylation during the leptotene to pachytene stages of meiosis.[12] HAT domains for this family are approximately 250 residues which include cysteine-rich, zinc binding domains as well as N-terminal chromodomains. The MYST proteins Esa1, Sas2 and Sas3 are found in yeast, MOF is found in Drosophila and mice while Tip60, MOZ, MORF, and HBO1 are found in humans.[9] Tip60 has roles in the regulation of gene transcription, HBO has been found to impact the DNA replication process, MORF is able to acetylate free histones (especially H3 and H4) as well as nucleosomal histones.[2]
p300/CBP family
Adenoviral E1A-associated protein of 300kDa (p300) and the CREB-binding protein (CBP) make up the next family of HATs.[10] This family of HATs contain HAT domains that are approximately 500 residues long and contain bromodomains as well as three cysteine-histidine rich domains that help with protein interactions.[9] These HATs are known to acetylate all of the histone subunits in the nucleosome. They also have the ability to acetylate and mediate non-histone proteins involved in transcription and are also involved in the cell-cycle, differentiation and apoptosis.[2]
Other HATs
There are other proteins that have acetylating abilities but differ in structure to the previously mentioned families. One HAT is called steroid receptor coactivator 1 (SRC1), which has a HAT domain located at the C-terminus end of the protein along with a basic helix-loop-helix and PAS A and PAS B domains with a LXXLL receptor interacting motif in the middle. Another is ATF-2 which contains a transcriptional activation (ACT) domain and a basic zipper DNA-binding (bZip) domain with a HAT domain in-between. The last is TAFII250 which has a Kinase domain at the N-terminus region, two bromodomains located at the C-terminus region and a HAT domain located in-between.[13]
Histone deacetylase (HDACs)
There are a total of four classes that categorize Histone Deacetylases (HDACs). Class I includes HDACs 1, 2, 3, and 8. Class II is divided into two subgroups, Class IIA and Class IIB. Class IIA includes HDACs 4, 5, 7, and 9 while Class IIB includes HDACs 6 and 10. Class III contains the Sirtuins and Class IV contains only HDAC11.[5][6] Classes of HDAC proteins are divided and grouped together based on the comparison to the sequence homologies of Rpd3, Hos1 and Hos2 for Class I HDACs, HDA1 and Hos3 for the Class II HDACs and the sirtuins for Class III HDACs.[6]
HDAC1 & HDAC2
HDAC1 & HDAC2 are in the first class of HDACs are most closely related to one another.[5][6] By analyzing the overall sequences of both HDACs, their similarity was found to be approximately 82% homologous.[5] These enzymes have been found to be inactive when isolated which led to the conclusion that they must be incorporated with cofactors in order to activate their deacetylase abilities.[5] There are three major protein complexes that HDAC 1 & 2 may incorporate themselves into. These complexes include Sin3 (named after its characteristic protein mSin3A), Nucleosome Remodelling and Deacetylating complex (NuRD), and Co-REST.[5][6] The Sin3 complex and the NuRD complex both contain HDACs 1 and 2, the Rb-associated protein 48 (RbAp48) and RbAp46 which make up the core of each complex.[2][6] Other complexes may be needed though in order to initiate the maximum amount of available activity possible. HDACs 1 and 2 can also bind directly to DNA binding proteins such as Yin and Yang 1 (YY1), Rb binding protein 1 and Sp1.[5] HDACs 1 and 2 have been found to express regulatory roles in key cell cycle genes including p21.[6]
Activity of these HDACs can be affected by phosphorylation. An increased amount of phosphorylation (hyperphosphorylation) leads to increased deacetylase activity, but degrades complex formation between HDACs 1 and 2 and between HDAC1 and mSin3A/YY1. A lower than normal amount of phosphorylation (hypophosphorylation) leads to a decrease in the amount of deacetylase activity, but increases the amount of complex formation. Mutation studies found that major phosphorylation happens at residues Ser421 and Ser423. Indeed, when these residues were mutated, a drastic reduction was seen in the amount of deacetylation activity.[5] This difference in the state of phosphorylation is a way of keeping an optimal level of phosphorylation to ensure there is no over or under expression of deacetylation. HDACs 1 and 2 have been found only exclusively in the nucleus.[2][6] In HDAC1 knockout (KO) mice, mice were found to die during embryogenesis and showed a drastic reduction in the production but increased expression of Cyclin-Dependent Kinase Inhibitors (CDKIs) p21 and p27. Not even upregulation of the other Class I HDACs could compensate for the loss of HDAC1. This inability to recover from HDAC1 KO leads researchers to believe that there are both functional uniqueness to each HDAC as well as regulatory cross-talk between factors.[6]
HDAC3
HDAC3 has been found to be most closely related to HDAC8. HDAC3 contains a non-conserved region in the C-terminal region that was found to be required for transcriptional repression as well as its deacetylase activity. It also contains two regions, one called a Nuclear Localization Signal (NLS) as well as a Nuclear Export Signal (NES). The NLS functions as a signal for nuclear action while an NES functions with HDACs that perform work outside of the nucleus. A presence of both signals for HDAC3 suggests it travels between the nucleus and the cytoplasm.[5] HDAC3 has even been found to interact with the plasma membrane.[6] Silencing Mediator for Retinoic Acid and Thyroid Hormone (SMRT) receptors and Nuclear Receptor Co-Repressor (N-CoR) factors must be utilized by HDAC3 in order to activate it.[5][6] Upon doing so, it gains the ability to co-precipitate with HDACs 4, 5, and 7. HDAC3 can also be found complexed together with HDAC-related protein (HDRP).[5] HDACs 1 and 3 have been found to mediate Rb-RbAp48 interactions which suggests that it functions in cell cycle progression.[5][6] HDAC3 also shows involvement in stem cell self-renewal and a transcription independent role in mitosis.[6]
HDAC8
HDAC8 has been found to be most similar to HDAC3. Its major feature is its catalytic domain which contains an NLS region in the center. Two transcripts of this HDAC have been found which include a 2.0kb transcript and a 2.4kb transcript.[5] Unlike the other HDAC molecules, when purified, this HDAC showed to be enzymatically active.[6] At this point, due to its recent discovery, it is not yet known if it is regulated by co-repressor protein complexes. Northern blots have revealed that different tissue types show varying degrees of HDAC8 expression[5] but has been observed in smooth muscles and is thought to contribute to contractility.[6]
Class IIA
The Class IIA HDACs includes HDAC4, HDAC5, HDAC7 and HDAC9. HDACs 4 and 5 have been found to most closely resemble each other while HDAC7 maintains a resemblance to both of them. There have been three discovered variants of HDAC9 including HDAC9a, HDAC9b and HDAC9c/HDRP, while more have been suspected. The variants of HDAC9 have been found to have similarities to the rest of the Class IIA HDACs. For HDAC9, the splicing variants can be seen as a way of creating a "fine-tuned mechanism" for differentiation expression levels in the cell. Different cell types may take advantage and utilize different isoforms of the HDAC9 enzyme allowing for different forms of regulation. HDACs 4, 5 and 7 have their catalytic domains located in the C-terminus along with an NLS region while HDAC9 has its catalytic domain located in the N-terminus. However, the HDAC9 variant HDAC9c/HDRP lacks a catalytic domain but has a 50% similarity to the N-terminus of HDACs 4 and 5.[5]
For HDACs 4, 5 and 7, conserved binding domains have been discovered that bind for C-terminal binding protein (CtBP), myocyte enhancer factor 2 (MEF2) and 14-3-3.[5][6] All three HDACs work to repress the myogenic transcription factor MEF2 which an essential role in muscle differentiation as a DNA binding transcription factor. Binding of HDACs to MEF2 inhibits muscle differentiation, which can be reversed by action of Ca2+/calmodulin-dependent kinase (CaMK) which works to dissociate the HDAC/MEF2 complex by phosphorylating the HDAC portion.[5] They have been seen to be involved in cellular hypertrophy in muscle control differentiation as well as cellular hypertrophy in muscle and cartilage tissues.[6] HDACs 5 and 7 have been shown to work in opposition to HDAC4 during muscle differentiation regulation so as to keep a proper level of expression. There has been evidence that these HDACs also interact with HDAC3 as a co-recruitment factor to the SMRT/N-CoR factors in the nucleus. Absence of the HDAC3 enzyme has shown to lead to inactivity which makes researchers believe that HDACs 4, 5 and 7 help the incorporation of DNA-binding recruiters for the HDAC3-containing HDAC complexes located in the nucleus.[5] When HDAC4 is knocked out in mice, they suffer from a pronounced chondrocyte hypertrophy and die due to extreme ossification. HDAC7 has been shown to suppress Nur77-dependent apoptosis. This interaction leads to a role in clonal expansion of T cells. HDAC9 KO mice are shown to suffer from cardiac hypertrophy which is exacerbated in mice that are double KO for HDACs 9 and 5.[6]
Class IIB
The Class IIB HDACs include HDAC6 and HDAC10. These two HDACs are most closely related to each other in overall sequence. However, HDAC6's catalytic domain is most similar to HDAC9.[5] A unique feature of HDAC6 is that it contains two catalytic domains in tandem of one another.[5][6] Another unique feature of HDAC6 is the HDAC6-, SP3, and Brap2-related zinc finger motif (HUB) domain in the C-terminus which shows some functions related to ubiquitination, meaning this HDAC is prone to degradation.[5] HDAC10 has two catalytic domains as well. One active domain is located in the N-terminus and a putative catalytic domain is located in the C-terminus[5][6] along with an NES domain.[5] Two putative Rb-binding domains have also been found on HDAC10 which shows it may have roles in the regulation of the cell cycle. Two variants of HDAC10 have been found, both having slight differences in length. HDAC6 is the only HDAC to be shown to act on tubulin, acting as a tubulin deacetylase which helps in the regulation of microtubule-dependent cell motility. It is mostly found in the cytoplasm but has been known to be found in the nucleus, complexed together with HDAC11. HDAC10 has been seen to act on HDACs 1, 2, 3 (or SMRT), 4, 5 and 7. Some evidence has been shown that it may have small interactions with HDAC6 as well. This leads researchers to believe that HDAC10 may function more as a recruiter rather than a factor for deacetylation. However, experiments conducted with HDAC10 did indeed show deacetylation activity.[5]
HDAC11
HDAC11 has been shown to be related to HDACs 3 and 8, but its overall sequence is quite different from the other HDACs, leading it to be in its own category.[5][6] HDAC11 has a catalytic domain located in its N-terminus. It has not been found incorporated in any HDAC complexes such as Nurd or SMRT which means it may have a special function unique to itself. It has been found that HDAC11 remains mainly in the nucleus.[5]
Biological functions
Transcription regulation
The discovery of histone acetylation causing changes in transcription activity can be traced back to the work of Vicent Allfrey and colleagues in 1964.[14] The group hypothesized that histone proteins modified by acetyl groups added negative charges to the positive lysines, and thus, reduced the interaction between DNA and histones.[15] Histone modification is now considered a major regulatory mechanism that is involved in many different stages of genetic functions.[16] Our current understanding is that acetylated lysine residues on histone tails is associated with transcriptional activation. In turn, deacetylated histones are associated with transcriptional repression. In addition, negative correlations have been found between several histone acetylation marks.[17]
The regulatory mechanism is thought to be twofold. Lysine is an amino acid with a positive charge when unmodified. Lysines on the amino terminal tails of histones have a tendency to weaken the chromatin's overall structure. Addition of an acetyl group, which carries a negative charge, effectively removes the positive charge and hence, reduces the interaction between the histone tail and the nucleosome.[18] This opens up the usually tightly packed nucleosome and allows transcription machinery to come into contact with the DNA template, leading to gene transcription.[1]: 242 Repression of gene transcription is achieved by the reverse of this mechanism. The acetyl group is removed by one of the HDAC enzymes during deacetylation, allowing histones to interact with DNA more tightly to form compacted nucleosome assembly. This increase in the rigid structure prevents the incorporation of transcriptional machinery, effectively silencing gene transcription.
Another implication of histone acetylation is to provide a platform for protein binding. As a posttranslational modification, the acetylation of histones can attract proteins to elongated chromatin that has been marked by acetyl groups. It has been hypothesized that the histone tails offer recognition sites that attract proteins responsible for transcriptional activation.[19] Unlike histone core proteins, histone tails are not part of the nucleosome core and are exposed to protein interaction. A model proposed that the acetylation of H3 histones activates gene transcription by attracting other transcription related complexes. Therefore, the acetyl mark provides a site for protein recognition where transcription factors interact with the acetylated histone tails via their bromodomain.[20]
Histone code hypothesis
The Histone code hypothesis suggests the idea that patterns of post-translational modifications on histones, collectively, can direct specific cellular functions.[21] Chemical modifications of histone proteins often occur on particular amino acids. This specific addition of single or multiple modifications on histone cores can be interpreted by transcription factors and complexes which leads to functional implications. This process is facilitated by enzymes such as HATs and HDACs that add or remove modifications on histones, and transcription factors that process and "read" the modification codes. The outcome can be activation of transcription or repression of a gene. For example, the combination of acetylation and phosphorylation have synergistic effects on the chromosomes overall structural condensation level and, hence, induces transcription activation of immediate early gene.[22]
Experiments investigating acetylation patterns of H4 histones suggested that these modification patterns are collectively maintained in mitosis and meiosis in order to modify long-term gene expression.[8] The acetylation pattern is regulated by HAT and HADC enzymes and, in turn, sets the local chromatin structure. In this way, acetylation patterns are transmitted and interconnected with protein binding ability and functions in subsequent cell generation.
Bromodomain
The bromodomain is a motif that is responsible for acetylated lysine recognition on histones by nucleosome remodelling proteins. Posttranslational modifications of N- and C-terminal histone tails attracts various transcription initiation factors that contain bromodomains, including human transcriptional coactivator PCAF, TAF1, GCN5 and CREB-binding protein (CBP), to the promoter and have a significance in regulating gene expression.[23] Structural analysis of transcription factors has shown that highly conserved bromodomains are essential for protein to bind to acetylated lysine. This suggests that specific histone site acetylation has a regulatory role in gene transcriptional activation.[24]
Human diseases
Inflammatory diseases
Gene expression is regulated by histone acetylation and deacetylation, and this regulation is also applicable to inflammatory genes. Inflammatory lung diseases are characterized by expression of specific inflammatory genes such as NF-κB and AP-1 transcription factor. Treatments with corticosteroids and theophylline for inflammatory lung diseases interfere with HAT/HDAC activity to turn off inflammatory genes.[25]
Specifically, gene expression data demonstrated increased activity of HAT and decreased level of HDAC activity in patients with Asthma.[26] Patients with chronic obstructive pulmonary disease showed there is an overall decrease in HDAC activity with unchanged levels of HAT activity.[27] Results have shown that there is an important role for HAT/HDAC activity balance in inflammatory lung diseases and provided insights on possible therapeutic targets.[28]
Cancer
Due to the regulatory role during transcription of epigenetic modifications in genes, it is not surprising that changes in epigenetic markers, such as acetylation, can contribute to cancer development. HDACs expression and activity in tumor cells is very different from normal cells. The overexpression and increased activity of HDACs has been shown to be characteristic of tumorigenesis and metastasis, suggesting an important regulatory role of histone deacetylation on the expression of tumor suppressor genes.[29] One of the examples is the regulation role of histone acetylation/deacetylation in P300 and CBP, both of which contribute to oncogenesis.[30]
Approved in 2006 by the U.S. Food and Drug Administration (FDA), Vorinostat represents a new category for anticancer drugs that are in development. Vorinostat targets histone acetylation mechanisms and can effectively inhibit abnormal chromatin remodeling in cancerous cells. Targets of Vorinostat includes HDAC1, HDAC2, HDAC3 and HDAC6.[31][32]
Carbon source availability is reflected in histone acetylation in cancer. Glucose and glutamine are the major carbon sources of most mammalian cells, and glucose metabolism is closely related to histone acetylation and deacetylation. Glucose availability affects the intracellular pool of acetyl-CoA, a central metabolic intermediate that is also the acetyl donor in histone acetylation. Glucose is converted to acetyl-CoA by the pyruvate dehydrogenase complex (PDC), which produces acetyl-CoA from glucose-derived pyruvate; and by adenosine triphosphate-citrate lyase (ACLY), which generates acetyl-CoA from glucose-derived citrate. PDC and ACLY activity depend on glucose availability, which thereby influences histone acetylation and consequently modulates gene expression and cell cycle progression. Dysregulation of ACLY and PDC contributes to metabolic reprogramming and promotes the development of multiple cancers. At the same time, glucose metabolism maintains the NAD+/NADH ratio, and NAD+ participates in SIRT-mediated histone deacetylation. SIRT enzyme activity is altered in various malignancies, and inhibiting SIRT6, a histone deacetylase that acts on acetylated H3K9 and H3K56, promotes tumorigenesis. SIRT7, which deacetylates H3K18 and thereby represses transcription of target genes, is activated in cancer to stabilize cells in the transformed state. Nutrients appear to modulate SIRT activity. For example, long-chain fatty acids activate the deacetylase function of SIRT6, and this may affect histone acetylation.[33]
Addiction
Epigenetic modifications of histone tails in specific regions of the brain are of central importance in addictions, and much of the work on addiction has focused on histone acetylation.[34][35][36] Once particular epigenetic alterations occur, they appear to be long lasting "molecular scars" that may account for the persistence of addictions.[34][37]
Cigarette smokers (about 21% of the US population[38]) are usually addicted to nicotine.[39] After 7 days of nicotine treatment of mice, acetylation of both histone H3 and histone H4 was increased at the FosB promoter in the nucleus accumbens of the brain, causing 61% increase in FosB expression.[40] This would also increase expression of the splice variant Delta FosB. In the nucleus accumbens of the brain, Delta FosB functions as a "sustained molecular switch" and "master control protein" in the development of an addiction.[41][42]
About 7% of the US population is addicted to alcohol. In rats exposed to alcohol for up to 5 days, there was an increase in histone 3 lysine 9 acetylation in the pronociceptin promoter in the brain amygdala complex. This acetylation is an activating mark for pronociceptin. The nociceptin/nociceptin opioid receptor system is involved in the reinforcing or conditioning effects of alcohol.[43]
Cocaine addiction occurs in about 0.5% of the US population. Repeated cocaine administration in mice induces hyperacetylation of histone 3 (H3) or histone 4 (H4) at 1,696 genes in one brain "reward" region [the nucleus accumbens (NAc)] and deacetylation at 206 genes.[44][45] At least 45 genes, shown in previous studies to be upregulated in the NAc of mice after chronic cocaine exposure, were found to be associated with hyperacetylation of H3 or H4. Many of these individual genes are directly related to aspects of addiction associated with cocaine exposure.[45][46]
In rodent models, many agents causing addiction, including tobacco smoke products,[47] alcohol,[48] cocaine,[49] heroin[50] and methamphetamine,[51][52] cause DNA damage in the brain. During repair of DNA damages some individual repair events may alter the acetylations of histones at the sites of damage, or cause other epigenetic alterations, and thus leave an epigenetic scar on chromatin.[37] Such epigenetic scars likely contribute to the persistent epigenetic changes found in addictions.
In 2013, 22.7 million persons aged 12 or older needed treatment for an illicit drug or alcohol use problem (8.6 percent of persons aged 12 or older).[38]
Other disorders
Suggested by the idea that the structure of chromatin can be modified to allow or deny access of transcription activators, regulatory functions of histone acetylation and deacetylation can have implications with genes that cause other diseases. Studies on histone modifications may reveal many novel therapeutic targets.
Based on different cardiac hypertrophy models, it has been demonstrated that cardiac stress can result in gene expression changes and alter cardiac function.[53] These changes are mediated through HATs/HDACs posttranslational modification signaling. HDAC inhibitor trichostatin A was reported to reduce stress induced cardiomyocyte autophagy.[54] Studies on p300 and CREB-binding protein linked cardiac hypertrophy with cellular HAT activity suggesting an essential role of histone acetylation status with hypertrophy responsive genes such as GATA4, SRF, and MEF2.[55][56][57][58]
Epigenetic modifications also play a role in neurological disorders. Deregulation of histones modification are found to be responsible for deregulated gene expression and hence associated with neurological and psychological disorders, such as Schizophrenia[59] and Huntington disease.[60] Current studies indicate that inhibitors of the HDAC family have therapeutic benefits in a wide range of neurological and psychiatric disorders.[61] Many neurological disorders only affect specific brain regions; therefore, understanding of the specificity of HDACs is still required for further investigations for improved treatments.
See also
References
- Watson JD, Baker TA, Gann A, Levine M, Losik R (2014). Molecular biology of the gene (Seventh ed.). Boston: Pearson/CSH Press. ISBN 978-0-321-76243-6.
- Verdone L, Agricola E, Caserta M, Di Mauro E (September 2006). "Histone acetylation in gene regulation". Briefings in Functional Genomics & Proteomics. 5 (3): 209–21. doi:10.1093/bfgp/ell028. PMID 16877467.
- Kuo MH, Allis CD (August 1998). "Roles of histone acetyltransferases and deacetylases in gene regulation". BioEssays. 20 (8): 615–26. doi:10.1002/(sici)1521-1878(199808)20:8<615::aid-bies4>3.0.co;2-h. PMID 9780836. S2CID 35433573.
- Grunstein M (September 1997). "Histone acetylation in chromatin structure and transcription" (PDF). Nature. 389 (6649): 349–52. Bibcode:1997Natur.389..349G. doi:10.1038/38664. PMID 9311776. S2CID 4419816.
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB (March 2003). "Histone deacetylases (HDACs): characterization of the classical HDAC family". The Biochemical Journal. 370 (Pt 3): 737–49. doi:10.1042/BJ20021321. PMC 1223209. PMID 12429021.
- Gallinari P, Di Marco S, Jones P, Pallaoro M, Steinkühler C (March 2007). "HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics". Cell Research. 17 (3): 195–211. doi:10.1038/sj.cr.7310149. PMID 17325692. S2CID 30268983.
- Struhl K (March 1998). "Histone acetylation and transcriptional regulatory mechanisms". Genes & Development. 12 (5): 599–606. doi:10.1101/gad.12.5.599. PMID 9499396.
- Turner BM (September 2000). "Histone acetylation and an epigenetic code". BioEssays. 22 (9): 836–45. doi:10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X. PMID 10944586.
- Marmorstein R (August 2001). "Structure of histone acetyltransferases". Journal of Molecular Biology. 311 (3): 433–44. doi:10.1006/jmbi.2001.4859. PMID 11492997.
- Yang XJ, Seto E (August 2007). "HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention". Oncogene. 26 (37): 5310–8. doi:10.1038/sj.onc.1210599. PMID 17694074. S2CID 10662910.
- Torok MS, Grant PA (2004). "Histone acetyltransferase proteins contribute to transcriptional processes at multiple levels". Advances in Protein Chemistry. 67: 181–99. doi:10.1016/S0065-3233(04)67007-0. ISBN 9780120342679. PMID 14969728.
- Jiang H, Gao Q, Zheng W, Yin S, Wang L, Zhong L, Ali A, Khan T, Hao Q, Fang H, Sun X, Xu P, Pandita TK, Jiang X, Shi Q (May 2018). "MOF influences meiotic expansion of H2AX phosphorylation and spermatogenesis in mice". PLOS Genetics. 14 (5): e1007300. doi:10.1371/journal.pgen.1007300. PMC 6019819. PMID 29795555.
- Marmorstein R, Roth SY (April 2001). "Histone acetyltransferases: function, structure, and catalysis". Current Opinion in Genetics & Development. 11 (2): 155–61. doi:10.1016/S0959-437X(00)00173-8. PMID 11250138.
- Allfrey VG, Faulkner R, Mirsky AE (May 1964). "Acetylation and Methylation of Histones and Their Possible Role in the Regulation of RNA Synthesis". Proceedings of the National Academy of Sciences of the United States of America. 51 (5): 786–94. Bibcode:1964PNAS...51..786A. doi:10.1073/pnas.51.5.786. PMC 300163. PMID 14172992.
- Mukhopadhyay R (2012). "Vincent Allfrey's Work on Histone Acetylation". Journal of Biological Chemistry. 287 (3): 2270–2271. doi:10.1074/jbc.O112.000248. PMC 3265906.
- Zentner GE, Henikoff S (March 2013). "Regulation of nucleosome dynamics by histone modifications". Nature Structural & Molecular Biology. 20 (3): 259–66. doi:10.1038/nsmb.2470. PMID 23463310. S2CID 23873925.
- Madrigal P, Krajewski P (July 2015). "Uncovering correlated variability in epigenomic datasets using the Karhunen-Loeve transform". BioData Mining. 8: 20. doi:10.1186/s13040-015-0051-7. PMC 4488123. PMID 26140054.
- Spange S, Wagner T, Heinzel T, Krämer OH (January 2009). "Acetylation of non-histone proteins modulates cellular signalling at multiple levels". The International Journal of Biochemistry & Cell Biology. 41 (1): 185–98. doi:10.1016/j.biocel.2008.08.027. PMID 18804549.
- Cheung P, Allis CD, Sassone-Corsi P (October 2000). "Signaling to chromatin through histone modifications". Cell. 103 (2): 263–71. doi:10.1016/s0092-8674(00)00118-5. PMID 11057899. S2CID 16237908.
- Winston F, Allis CD (July 1999). "The bromodomain: a chromatin-targeting module?". Nature Structural Biology. 6 (7): 601–4. doi:10.1038/10640. PMID 10404206. S2CID 22196542.
- Chi P, Allis CD, Wang GG (July 2010). "Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers". Nature Reviews. Cancer. 10 (7): 457–69. doi:10.1038/nrc2876. PMC 3262678. PMID 20574448.
- Barratt MJ, Hazzalin CA, Cano E, Mahadevan LC (May 1994). "Mitogen-stimulated phosphorylation of histone H3 is targeted to a small hyperacetylation-sensitive fraction". Proceedings of the National Academy of Sciences of the United States of America. 91 (11): 4781–5. Bibcode:1994PNAS...91.4781B. doi:10.1073/pnas.91.11.4781. PMC 43872. PMID 8197135.
- Sanchez R, Zhou MM (September 2009). "The role of human bromodomains in chromatin biology and gene transcription". Current Opinion in Drug Discovery & Development. 12 (5): 659–65. PMC 2921942. PMID 19736624.
- Filippakopoulos P, Knapp S (May 2014). "Targeting bromodomains: epigenetic readers of lysine acetylation". Nature Reviews. Drug Discovery. 13 (5): 337–56. doi:10.1038/nrd4286. PMID 24751816. S2CID 12172346.
- Barnes PJ, Adcock IM, Ito K (March 2005). "Histone acetylation and deacetylation: importance in inflammatory lung diseases". The European Respiratory Journal. 25 (3): 552–63. doi:10.1183/09031936.05.00117504. PMID 15738302.
- Kuo CH, Hsieh CC, Lee MS, Chang KT, Kuo HF, Hung CH (January 2014). "Epigenetic regulation in allergic diseases and related studies". Asia Pacific Allergy. 4 (1): 14–8. doi:10.5415/apallergy.2014.4.1.14. PMC 3921865. PMID 24527405.
- Mroz RM, Noparlik J, Chyczewska E, Braszko JJ, Holownia A (November 2007). "Molecular basis of chronic inflammation in lung diseases: new therapeutic approach". Journal of Physiology and Pharmacology. 58 Suppl 5 (Pt 2): 453–60. PMID 18204158.
- Barnes PJ, Adcock IM, Ito K (March 2005). "Histone acetylation and deacetylation: importance in inflammatory lung diseases". The European Respiratory Journal. 25 (3): 552–63. doi:10.1183/09031936.05.00117504. PMID 15738302.
- Glozak MA, Seto E (August 2007). "Histone deacetylases and cancer". Oncogene. 26 (37): 5420–32. doi:10.1038/sj.onc.1210610. PMID 17694083.
- Cohen I, Poręba E, Kamieniarz K, Schneider R (June 2011). "Histone modifiers in cancer: friends or foes?". Genes & Cancer. 2 (6): 631–47. doi:10.1177/1947601911417176. PMC 3174261. PMID 21941619.
- Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM, Frankel SR (January 2007). "Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL)". Blood. 109 (1): 31–9. doi:10.1182/blood-2006-06-025999. PMC 1785068. PMID 16960145.
- Grant S, Easley C, Kirkpatrick P (January 2007). "Vorinostat". Nature Reviews. Drug Discovery. 6 (1): 21–2. doi:10.1038/nrd2227. PMID 17269160.
- Wang YP, Lei QY (May 2018). "Metabolic recoding of epigenetics in cancer". Cancer Communications. 38 (1): 25. doi:10.1186/s40880-018-0302-3. PMC 5993135. PMID 29784032.
- Robison AJ, Nestler EJ (October 2011). "Transcriptional and epigenetic mechanisms of addiction". Nature Reviews. Neuroscience. 12 (11): 623–37. doi:10.1038/nrn3111. PMC 3272277. PMID 21989194.
- Hitchcock LN, Lattal KM (2014). "Histone-mediated epigenetics in addiction". Epigenetics and Neuroplasticity—Evidence and Debate. pp. 51–87. doi:10.1016/B978-0-12-800977-2.00003-6. ISBN 9780128009772. PMC 5914502. PMID 25410541.
{{cite book}}:|journal=ignored (help) - McQuown SC, Wood MA (April 2010). "Epigenetic regulation in substance use disorders". Current Psychiatry Reports. 12 (2): 145–53. doi:10.1007/s11920-010-0099-5. PMC 2847696. PMID 20425300.
- Dabin J, Fortuny A, Polo SE (June 2016). "Epigenome Maintenance in Response to DNA Damage". Molecular Cell. 62 (5): 712–27. doi:10.1016/j.molcel.2016.04.006. PMC 5476208. PMID 27259203.
- Substance Abuse and Mental Health Services Administration, Results from the 2013 National Survey on Drug Use and Health: Summary of National Findings, NSDUH Series H-48, HHS Publication No. (SMA) 14-4863. Rockville, MD: Substance Abuse and Mental Health Services Administration, 2014
- "Is nicotine addictive?".
- Levine A, Huang Y, Drisaldi B, Griffin EA, Pollak DD, Xu S, Yin D, Schaffran C, Kandel DB, Kandel ER (November 2011). "Molecular mechanism for a gateway drug: epigenetic changes initiated by nicotine prime gene expression by cocaine". Science Translational Medicine. 3 (107): 107ra109. doi:10.1126/scitranslmed.3003062. PMC 4042673. PMID 22049069.
- Ruffle JK (November 2014). "Molecular neurobiology of addiction: what's all the (Δ)FosB about?". The American Journal of Drug and Alcohol Abuse. 40 (6): 428–37. doi:10.3109/00952990.2014.933840. PMID 25083822. S2CID 19157711.
- Nestler EJ, Barrot M, Self DW (September 2001). "DeltaFosB: a sustained molecular switch for addiction". Proceedings of the National Academy of Sciences of the United States of America. 98 (20): 11042–6. Bibcode:2001PNAS...9811042N. doi:10.1073/pnas.191352698. PMC 58680. PMID 11572966.
- D'Addario C, Caputi FF, Ekström TJ, Di Benedetto M, Maccarrone M, Romualdi P, Candeletti S (February 2013). "Ethanol induces epigenetic modulation of prodynorphin and pronociceptin gene expression in the rat amygdala complex". Journal of Molecular Neuroscience. 49 (2): 312–9. doi:10.1007/s12031-012-9829-y. PMID 22684622. S2CID 14013417.
- Walker DM, Nestler EJ (2018). "Neuroepigenetics and addiction". Neurogenetics, Part II. pp. 747–765. doi:10.1016/B978-0-444-64076-5.00048-X. ISBN 9780444640765. PMC 5868351. PMID 29478612.
{{cite book}}:|journal=ignored (help) - Renthal W, Kumar A, Xiao G, Wilkinson M, Covington HE, Maze I, Sikder D, Robison AJ, LaPlant Q, Dietz DM, Russo SJ, Vialou V, Chakravarty S, Kodadek TJ, Stack A, Kabbaj M, Nestler EJ (May 2009). "Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins". Neuron. 62 (3): 335–48. doi:10.1016/j.neuron.2009.03.026. PMC 2779727. PMID 19447090.
- https://www.drugsandalcohol.ie/12728/1/NIDA_Cocaine.pdf
- Adhami N, Chen Y, Martins-Green M (October 2017). "Biomarkers of disease can be detected in mice as early as 4 weeks after initiation of exposure to third-hand smoke levels equivalent to those found in homes of smokers". Clinical Science. 131 (19): 2409–2426. doi:10.1042/CS20171053. PMID 28912356.
- Rulten SL, Hodder E, Ripley TL, Stephens DN, Mayne LV (July 2008). "Alcohol induces DNA damage and the Fanconi anemia D2 protein implicating FANCD2 in the DNA damage response pathways in brain". Alcoholism: Clinical and Experimental Research. 32 (7): 1186–96. doi:10.1111/j.1530-0277.2008.00673.x. PMID 18482162.
- de Souza MF, Gonçales TA, Steinmetz A, Moura DJ, Saffi J, Gomez R, Barros HM (April 2014). "Cocaine induces DNA damage in distinct brain areas of female rats under different hormonal conditions". Clinical and Experimental Pharmacology & Physiology. 41 (4): 265–9. doi:10.1111/1440-1681.12218. PMID 24552452. S2CID 20849951.
- Qiusheng Z, Yuntao Z, Rongliang Z, Dean G, Changling L (July 2005). "Effects of verbascoside and luteolin on oxidative damage in brain of heroin treated mice". Die Pharmazie. 60 (7): 539–43. PMID 16076083.
- Johnson Z, Venters J, Guarraci FA, Zewail-Foote M (June 2015). "Methamphetamine induces DNA damage in specific regions of the female rat brain". Clinical and Experimental Pharmacology & Physiology. 42 (6): 570–5. doi:10.1111/1440-1681.12404. PMID 25867833. S2CID 24182756.
- Tokunaga I, Ishigami A, Kubo S, Gotohda T, Kitamura O (August 2008). "The peroxidative DNA damage and apoptosis in methamphetamine-treated rat brain". The Journal of Medical Investigation. 55 (3–4): 241–5. doi:10.2152/jmi.55.241. PMID 18797138.
- Mano H (January 2008). "Epigenetic abnormalities in cardiac hypertrophy and heart failure". Environmental Health and Preventive Medicine. 13 (1): 25–9. doi:10.1007/s12199-007-0007-8. PMC 2698246. PMID 19568876.
- Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, Rothermel BA, Gillette TG, Hill JA (March 2011). "Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy". Proceedings of the National Academy of Sciences of the United States of America. 108 (10): 4123–8. Bibcode:2011PNAS..108.4123C. doi:10.1073/pnas.1015081108. PMC 3053983. PMID 21367693.
- Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN (August 2002). "Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy". Cell. 110 (4): 479–88. doi:10.1016/S0092-8674(02)00861-9. PMC 4459650. PMID 12202037.
- Lehmann LH, Worst BC, Stanmore DA, Backs J (May 2014). "Histone deacetylase signaling in cardioprotection". Cellular and Molecular Life Sciences. 71 (9): 1673–90. doi:10.1007/s00018-013-1516-9. PMC 3983897. PMID 24310814.
- Wang Y, Miao X, Liu Y, Li F, Liu Q, Sun J, Cai L (2014). "Dysregulation of histone acetyltransferases and deacetylases in cardiovascular diseases". Oxidative Medicine and Cellular Longevity. 2014: 641979. doi:10.1155/2014/641979. PMC 3945289. PMID 24693336.
- Shikama N, Lutz W, Kretzschmar R, Sauter N, Roth JF, Marino S, Wittwer J, Scheidweiler A, Eckner R (October 2003). "Essential function of p300 acetyltransferase activity in heart, lung and small intestine formation". The EMBO Journal. 22 (19): 5175–85. doi:10.1093/emboj/cdg502. PMC 204485. PMID 14517255.
- Tang B, Dean B, Thomas EA (December 2011). "Disease- and age-related changes in histone acetylation at gene promoters in psychiatric disorders". Translational Psychiatry. 1 (12): e64. doi:10.1038/tp.2011.61. PMC 3305989. PMID 22832356.
- Lee J, Hwang YJ, Kim KY, Kowall NW, Ryu H (October 2013). "Epigenetic mechanisms of neurodegeneration in Huntington's disease". Neurotherapeutics. 10 (4): 664–76. doi:10.1007/s13311-013-0206-5. PMC 3805871. PMID 24006238.
- Grayson DR, Kundakovic M, Sharma RP (February 2010). "Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders?". Molecular Pharmacology. 77 (2): 126–35. doi:10.1124/mol.109.061333. PMID 19917878. S2CID 3112549.