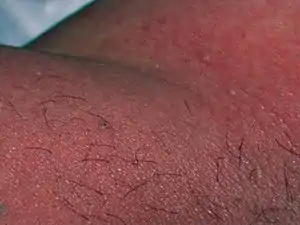
Acute generalized exanthematous pustulosis
Acute generalized exanthematous pustulosis (AGEP) (also known as pustular drug eruption and toxic pustuloderma) is a rare skin reaction that in 90% of cases is related to medication.
| Acute generalized exanthematous pustulosis | |
|---|---|
| Other names | Toxic pustuloderma, pustular drug eruption |
 | |
| Acute generalized exanthematous pustulosis | |
| Specialty | Dermatology |
AGEP is characterized by sudden skin eruptions that appear on average five days after a medication is started. These eruptions are pustules, i.e. small red white or red elevations of the skin that contain cloudy or purulent material (pus).[1] The skin lesions usually resolve within 1–3 days of stopping the offending medication.[2] However, more severe cases are associated with a more persistent disorder that may be complicated by secondary skin infections and/or involvement of the liver, lung, and/or kidney.[3]
Severe cutaneous adverse reaction (SCAR) disorders are regarded as the drug-induced activation of T cells which then initiate innate immune responses that are inappropriately directed against self tissues. Studies on the DRESS syndrome, Stevens–Johnson syndrome (SJS), toxic epidermal necrolysis (TEN) and SJS/TEN overlap indicate that many individuals are predisposed to develop these reactions to a particular medication based on their genetically determined expression of particular human leukocyte antigen (i.e. HLA) alleles or T-cell receptors and/or their efficiencies in adsorbing, distributing to tissues, metabolizing, and/or eliminating) a particular SCARS-inducing medication. Evidence for these predispositions in AGEP has not been as well-established.[2][4][5]
Signs and symptoms
AGEP is an acute drug eruption characterized by numerous small, primarily non-follicular, sterile skin pustules arising within large areas of red swollen skin usually within days of taking an inciting drug.[6] The skin eruptions are often pruritic and accompanied by fever, headache, a high number of neutrophils and eosinophils in the blood, and elevated blood levels of markers for inflammation (i.e. erythrocyte sedimentation rate and C-reactive protein). The skin eruptions typically end within a week after causative drug is discontinued.[3]
Rare cases of lung and bone marrow involvement have also been reported to complicate AGEP.[3][7] However, involvement of these organs typically resolve along with the skin eruptions. AGEP typically shows a mild course: usually, it is not associated with life-threatening complicates although superinfections of skin lesions may be serious or even life-threatening. AGEP has a mortality rate of less than 5%.[2][7][8]
Cause
About 90% of AGEP reactions are associated with medications. The remaining cases of AGEP have been associated with infective and other agents.[7]
Medicines
The most frequently reported drugs that have been associated with the development of AGEP include penicillin, aminopenicillins, macrolides, quinolones, sulfonamides, hydroxychloroquine, terbinafine, and diltiazem.[7] A more complete list of drugs sorted by their intended actions are:[3][7][9][10][11]
- Antibiotics: Penicillin, ampicillin, amoxicillin, clindamycin, cephalexin, cefepime, cefoxitin, cefazolin, various sulfonamides, various quinolones, vancomycin, levofloxacin, imipenem, meropenem, pristinamycin
- Antifungals: Terbinafine, ketoconazole, fluconazole
- Anti-inflammatories: Aspirin, celecoxib
- Other agents: Hydroxychloroquine (an antimalarial agent), diltiazem (a calcium channel blocker), omeprazole (a proton-pump inhibitor), clenbuterol (a decongestant, bronchodilator), hydroxyzine[12] (An antihistamine medication), and clopidogrel (an indirectly acting platelet inhibitor)
Microbe infections
Infections with Parvovirus B19, mycoplasma, cytomegalovirus, coxsackie B4 virus, Epstein-Barr virus (EBV),[13][14] Chlamydophila pneumoniae, E. coli, and Echinococcus have been reported to be associated with the development of AGEP in the absence of an apparent drug-induced cause. The pathophysiology for the development of these drug-independent cases of AGEP is unclear.[7] Viral infections have also been observed to be associated with the development of SJS, SJS/TEN, and TEN in the absence of a causative drug.[7][10][15]
Other agents
Herbal medications, spider bites, iopamidol (used for radiocontrast), lacquers, mercury, psoralen (combined with ultraviolet A to treat psoriasis), and xenobiotics have been associated with the development of AGEP in case reports.[11]
Pathophysiology
Like other drug-induced SCARs disorders, AGEP is a type IV hypersensitivity reaction in which a drug or its metabolite stimulates cytotoxic T cells (i.e. CD8+ T cells) or T helper cells (i.e. CD4+ T cells) to initiate autoimmune reactions that attack self tissues. SCARs are type IV, subtype IVb (DRESS syndrome), type IV, subtype IVc (SJS, SJS/TEN, TEN), or type IV, subtype IVd (AGEP) hypersensitivity reactions. AGEP therefore differs from the other SCARs disorders in that it involves the tissue-injuring action of inappropriately activated neutrophils and the excessive production of cytokines which stimulate production of, recruit to tissues, and/or activation of neutrophils (Interleukin 8, Interleukin 17, GM-CSF) and promote innate immune and autoimmune responses (Interleukin 22).[2][7][8]
AGEP also differs from the other SCARs disorders in respect to the level of evidence supporting the underlying mechanism by which a drug or its metabolite stimulates CD8+ T or CD4+ T cells. Studies indicate that the mechanism by which a drug or its metabolites accomplishes this stimulation involves subverting the antigen presentation pathways of the innate immune system. A drug or metabolite covalently binds with a host protein to form a non-self, drug-related epitope. An antigen-presenting cell (APC) takes up these proteins; digests them into small peptides; places the peptides in a groove on the human leukocyte antigen (i.e. HLA) component of their major histocompatibility complex (i.e. MHC) (APC); and presents the MHC-associated peptides to the T-cell receptor on CD8+ T or CD4+ T cells. Those peptides expressing a drug-related, non-self epitope on their HLA-A, HLA-B, HLA-C, HLA-DM, HLA-DO, HLA-DP, HLA-DQ, or HLA-DR proteins may bind to a T-cell receptor to stimulate the receptor-bearing parent T cell to attack self tissues. Alternatively, a drug or metabolite may also stimulate T cells by inserting into the groove on a HLA protein to serve as a non-self epitope, bind outside of this groove to alter a HLA protein so that it forms a non-self epitope, or bypass the APC by binding directly to a T cell receptor. However, non-self epitopes must bind to specific HLA serotypes to stimulate T cells and the human population expresses some 13,000 different HLA serotypes while an individual expresses only a fraction of them. Since a SCARs-inducing drug or metabolite interacts with only one or a few HLA serotypes, their ability to induce SCARs is limited to those individuals who express HLA serotypes targeted by the drug or metabolite.[5][8] Thus, only rare individuals are predisposed to develop SCARs in response to a particular drug on the basis of their expression of HLA serotypes.[16] Studies have identified several HLA serotypes associated with development of DRESS syndrome, SJS, SJS/TEN, and TEN in response to various drugs which elici these disorders, developed tests to identify individuals who express these serotypes, and thereby determined that these individuals should avoid the offending drug. HLA serotypes associated with AGEP and specific drugs have not been identified.[5] A study conducted in 1995 identified of HLA-B51, HLA-DR11, and HLA-DQ3 of unknown serotypes to be associated with development of AGEP but the results have not been confirmed, expanded to identify the serotypes involved, nor therefore useful in identifying individuals predisposed to develop AGEP in response to any drug.[2] Similarly, a specific T cell receptor variant has been associated with the development of DRESS syndrome, SJS, SJS/TEN, and TEN but not AGEP.[2][17]
Variations in ADME, i.e. an individual's efficiency in absorbing, distributing, metabolizing, and excreting a drug) has been found to occur in cases of the DRESS syndrome, SJS, SJS/TEN, and TEN. These variations influence the levels and duration of a drug or drug metabolite in tissues and thereby impact the drug's or drug metabolite's ability to evoke SCARs.[18]
In rare cases, the development of AGEP has been reported to occur in individuals with loss of function mutations in their IL36RN gene. This gene codes for the interleukin 36 receptor antagonist (IL36RA). IL36RA blocks the pro-inflammatory actions of Interleukin-36 cytokines (viz., IL-36α, IL-36β and IL-36γ) on keratinocytes, synoviocytes, dendritic cells, macrophages, and T cells. It does so by binding to, but not stimulating, these cytokines' receptors, IL1RL2 and IL1RAP, thereby interfering with the interleukin-36 cytokines' binding to and stimulating IL1RL2 and IL1RAP. However, the IL36RN loss of function mutation has been reported in cases of generalized pustular psoriasis.[3][19] The presence of this mutation in two seemingly unrelated disorders has led to suggestions that the classification of AGEP as a SCARs or a form of psoriasis requires study.[19]
Diagnosis
The diagnosis of AGEP may be forthright in typical cases in which an individual: has taken a drug known to cause the disorder; develops multiple sterile pustules overlying large areas of red swollen skin starting a few days after initial drug intake; and has a histology of biopsied lesions that shows pustules just below the skin's Stratum corneum (outermost layer), apoptotic (i.e. necrotic) keratinocytes, spongiosis of the stratum spinosum, and infiltration of these tissues by neutrophils plus, in many but not all cases, eosinophils.[7] Many cases of AGEP, however, present less clear cut clinical features of the disorder. AGEP must be differentiated from generalized pustular psoriasis (GPP) with which it shares many clinical and histological features. A history of psoriasis, the presence of typical psoriatic skin lesions at the time of diagnosis, and histological evidence in skin lesions of necrotic keratinocytes, neutrophil-rich infiltrates, eosinophil infiltrates, and/or lack of tortuous or dilated blood vessels favors a diagnosis of to AGEP.[20] Other conditions sometimes confused with AGEP include pustular eruptions caused by bacteria, funguses, herpesviridae, and the varicella zoster virus (i.e. causative agent of chicken pox).
Several tests have been proposed to be useful for supporting the diagnosis of and/or implicating a particular drug as the cause of AGEP particularly in individuals who develop skin lesions while taking multiple drugs. These include patch tests in which small amounts of suspect drugs absorbed on patches are applied to the skin; skin allergy tests in which drugs are applied by skin prick or intradermal injection; and oral provocation in which drugs are taken in a single small dose orally. These tests have not been widely adopted because of their insensitivity and, most particular with oral provocation tests, the possibility of causing a relapse or worsening or the disorder. In vitro tests, including mixed lymphocyte reaction tests in which the response of individuals' blood mononuclear cells to suspect drugs and ELISPOT tests in which specific drug-reactive lymphocytes or their drug-induced release of AGEP mediators (e.g. interferon-γ interleukin 4, or granulysin) are measured have likewise not been broadly adopted because of their lack of specificity.[8]
Classification
The disorder is classified in the group of severe cutaneous adverse reactions (i.e. SCARs). The SCARs group of disorders includes four other drug-induced skin reactions: drug reaction with eosinophilia and systemic symptoms (DRESS syndrome), Stevens–Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and Stevens–Johnson/toxic epidermal necrolysis overlap syndrome (SJS/TEN). SJS, SJS/TEN, and TEN, while initially described as distinct adverse drug-induced cutaneous reactions are now regarded as manifestations of epidermal necrolysis differing only in extent of skin involvement. While all of five SCARs disorders are potentially lethal, AGEP has the lowest mortality of the group.[2][7]
Treatment
The treatment of AGEP begins with the immediate cessation of the offending drug. For individuals developing AGEP while taking multiple drugs, non-essential drugs should be discontinued and essential drugs should be replaced by chemically unrelated drugs that are used as alternatives to the discontinued drug(s). In cases of multiple drug intake, skin and/or in vitro testing may be of some use in identifying the offending drug. Beyond identifying and discontinuing the offending drug, individuals with mild symptoms may require no further treatment. Those troubled by more significant symptoms such as itching or fever may require antihistamines, topical corticosteroids, systemic corticosteroids, and/or antipyretics. Individuals with liver, lung, kidney, and/or severe skin complications may require high dosage systemic corticosteroids and organ-specific interventions. Skin infections, which may lead to sepsis, are potentially lethal complications of AGEP; preventative methods and rapid treatment of such infections with appropriate antibiotics and, where needed, further supportive measures are critical in the treatment of this complication. Overall, however, AGEP has a lethality of less than 5% with recent reports showing no fatalities. Typically, individuals with AGEP have rapid rates of recovery even in when experiencing the cited complications.[3][8][9][21]
See also
References
- James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0-7216-2921-0.: 124
- Hoetzenecker W, Nägeli M, Mehra ET, Jensen AN, Saulite I, Schmid-Grendelmeier P, Guenova E, Cozzio A, French LE (2016). "Adverse cutaneous drug eruptions: current understanding". Seminars in Immunopathology. 38 (1): 75–86. doi:10.1007/s00281-015-0540-2. PMID 26553194. S2CID 333724.
- Alniemi DT, Wetter DA, Bridges AG, El-Azhary RA, Davis MD, Camilleri MJ, McEvoy MT (2017). "Acute generalized exanthematous pustulosis: clinical characteristics, etiologic associations, treatments, and outcomes in a series of 28 patients at Mayo Clinic, 1996-2013". International Journal of Dermatology. 56 (4): 405–414. doi:10.1111/ijd.13434. PMID 28084022. S2CID 21325754.
- Halevy S (August 2009). "Acute generalized exanthematous pustulosis". Curr Opin Allergy Clin Immunol. 9 (4): 322–8. doi:10.1097/ACI.0b013e32832cf64e. PMID 19458527. S2CID 205434868.
- Garon SL, Pavlos RK, White KD, Brown NJ, Stone CA, Phillips EJ (2017). "Pharmacogenomics of off-target adverse drug reactions". British Journal of Clinical Pharmacology. 83 (9): 1896–1911. doi:10.1111/bcp.13294. PMC 5555876. PMID 28345177.
- Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. pp. 297, 303, 308, 309, 470. ISBN 978-1-4160-2999-1.
- Feldmeyer L, Heidemeyer K, Yawalkar N (July 2016). "Acute Generalized Exanthematous Pustulosis: Pathogenesis, Genetic Background, Clinical Variants and Therapy". International Journal of Molecular Sciences. 17 (8): 1214. doi:10.3390/ijms17081214. PMC 5000612. PMID 27472323.
- Duong TA, Valeyrie-Allanore L, Wolkenstein P, Chosidow O (2017). "Severe cutaneous adverse reactions to drugs". Lancet. 390 (10106): 1996–2011. doi:10.1016/S0140-6736(16)30378-6. PMID 28476287. S2CID 9506967.
- Thienvibul C, Vachiramon V, Chanprapaph K (2015). "Five-Year Retrospective Review of Acute Generalized Exanthematous Pustulosis". Dermatology Research and Practice. 2015: 260928. doi:10.1155/2015/260928. PMC 4689982. PMID 26783390.
- Szatkowski J, Schwartz RA (November 2015). "Acute generalized exanthematous pustulosis (AGEP): A review and update". Journal of the American Academy of Dermatology. 73 (5): 843–8. doi:10.1016/j.jaad.2015.07.017. PMID 26354880.
- Choi MJ, Kim HS, Park HJ, Park CJ, Lee JD, Lee JY, Kim HO, Park YM (May 2010). "Clinicopathologic manifestations of 36 Korean patients with acute generalized exanthematous pustulosis: a case series and review of the literature". Annals of Dermatology. 22 (2): 163–9. doi:10.5021/ad.2010.22.2.163. PMC 2883418. PMID 20548906.
- "HYDROXYZINE HYDROCHLORIDE tablet". Daily Med. U.S. National Library of Medicine. Retrieved 25 October 2020.
- Ropars, Nolwenn; Darrieux, Laure; Tisseau, Laurent; Safa, Gilles (2014-09-28). "Acute generalized exanthematous pustulosis associated with primary Epstein-Barr virus infection". JAAD Case Reports. 1 (1): 9–11. doi:10.1016/j.jdcr.2014.09.004. ISSN 2352-5126. PMC 4802527. PMID 27075126.
- "Acute generalised exanthematous pustulosis | DermNet NZ". dermnetnz.org. Retrieved 2021-01-14.
- Lerch M, Mainetti C, Terziroli Beretta-Piccoli B, Harr T (February 2018). "Current Perspectives on Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis". Clinical Reviews in Allergy & Immunology. 54 (1): 147–176. doi:10.1007/s12016-017-8654-z. PMID 29188475. S2CID 46796285.
- Pichler WJ, Hausmann O (2016). "Classification of Drug Hypersensitivity into Allergic, p-i, and Pseudo-Allergic Forms". International Archives of Allergy and Immunology. 171 (3–4): 166–179. doi:10.1159/000453265. PMID 27960170.
- Wang CW, Dao RL, Chung WH (2016). "Immunopathogenesis and risk factors for allopurinol severe cutaneous adverse reactions". Current Opinion in Allergy and Clinical Immunology. 16 (4): 339–45. doi:10.1097/ACI.0000000000000286. PMID 27362322. S2CID 9183824.
- Adler NR, Aung AK, Ergen EN, Trubiano J, Goh MS, Phillips EJ (November 2017). "Recent advances in the understanding of severe cutaneous adverse reactions". The British Journal of Dermatology. 177 (5): 1234–1247. doi:10.1111/bjd.15423. PMC 5582023. PMID 28256714.
- Bachelez H (January 2018). "Pustular psoriasis and related pustular skin diseases". The British Journal of Dermatology. 178 (3): 614–618. doi:10.1111/bjd.16232. PMID 29333670. S2CID 4436573.
- Orime M (2017). "Immunohistopathological Findings of Severe Cutaneous Adverse Drug Reactions". Journal of Immunology Research. 2017: 6928363. doi:10.1155/2017/6928363. PMC 5684554. PMID 29226159.
- Canadian Medical Association Journal, September 15, 2009, pp 393-396