Marfan syndrome
Marfan syndrome (MFS) is a multi-systemic genetic disorder that affects the connective tissue.[5][6][1] Those with the condition tend to be tall and thin, with long arms, legs, fingers, and toes.[1] They also typically have exceptionally flexible joints and abnormally curved spines.[1] The most serious complications involve the heart and aorta, with an increased risk of mitral valve prolapse and aortic aneurysm.[1][7] The lungs, eyes, bones, and the covering of the spinal cord are also commonly affected.[1] The severity of the symptoms is variable.[1]
| Marfan syndrome | |
|---|---|
| Other names | Marfan's syndrome |
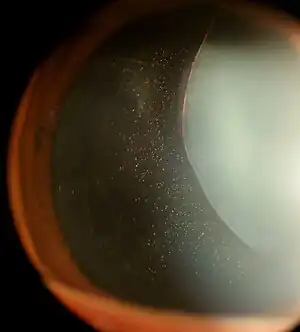 | |
| Ectopia lentis in Marfan syndrome: Zonular fibers are seen. | |
| Specialty | Medical genetics |
| Symptoms | Tall thin build; long arms, legs and fingers; flexible fingers and toes[1] |
| Complications | Scoliosis, mitral valve prolapse, aortic aneurysm[1] |
| Duration | Long term[1] |
| Causes | Genetic (autosomal dominant)[1] |
| Diagnostic method | Ghent criteria[2] |
| Differential diagnosis | Loeys-Dietz syndrome, Ehlers-Danlos syndrome |
| Medication | Beta blockers, calcium channel blockers, ACE inhibitors[3][4] |
| Prognosis | Often normal life expectancy[1] |
| Frequency | 1 in 5,000–10,000[3] |
MFS is caused by a mutation in FBN1, one of the genes that make fibrillin, which results in abnormal connective tissue.[1] It is an autosomal dominant disorder.[1] In about 75% of cases, it is inherited from a parent with the condition, while in about 25% it is a new mutation.[1] Diagnosis is often based on the Ghent criteria.[2][3]
There is no known cure for MFS.[1] Many of those with the disorder have a normal life expectancy with proper treatment.[1] Management often includes the use of beta blockers such as propranolol or atenolol or, if they are not tolerated, calcium channel blockers or ACE inhibitors.[3][4] Surgery may be required to repair the aorta or replace a heart valve.[4] Avoiding strenuous exercise is recommended for those with the condition.[3]
About 1 in 5,000 to 1 in 10,000 people have MFS.[3][8] Rates of the condition are similar in different regions of the world.[8] It is named after French pediatrician Antoine Marfan, who first described it in 1896.[9][10]
Signs and symptoms
More than 30 signs and symptoms are variably associated with Marfan syndrome. The most prominent of these affect the skeletal, cardiovascular, and ocular systems, but all fibrous connective tissue throughout the body can be affected.
Skeletal system
Most of the readily visible signs are associated with the skeletal system. Many people with Marfan syndrome grow to above-average height, and some have disproportionately long, slender limbs with thin, weak wrists and long fingers and toes.
The Steinberg sign, also known as the thumb sign, is one of the clinical examination tests for Marfan disease in the hands. It is a clinical test in which the tip of the thumb extends beyond the palm when the thumb is clasped in the clenched hand.[11][12][13]
Besides affecting height and limb proportions, people with Marfan syndrome may have abnormal lateral curvature of the spine scoliosis, thoracic lordosis, abnormal indentation (pectus excavatum) or protrusion (pectus carinatum) of the sternum, abnormal joint flexibility, a high-arched palate with crowded teeth and an overbite, flat feet, hammer toes, stooped shoulders, and unexplained stretch marks on the skin. It can also cause pain in the joints, bones, and muscles. Some people with Marfan have speech disorders resulting from symptomatic high palates and small jaws. Early osteoarthritis may occur. Other signs include limited range of motion in the hips due to the femoral head protruding into abnormally deep hip sockets.[14][15]
Eyes
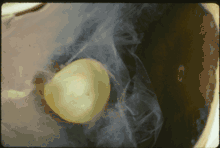
In Marfan syndrome, the health of the eye can be affected in many ways, but the principal change is partial lens dislocation, where the lens is shifted out of its normal position.[15] This occurs because of weakness in the ciliary zonules, the connective tissue strands which suspend the lens within the eye. The mutations responsible for Marfan syndrome weaken the zonules and cause them to stretch. The inferior zonules are most frequently stretched resulting in the lens shifting upwards and outwards, but it can shift in other directions as well. Nearsightedness (myopia), and blurred vision are common due to connective tissue defects in the eye.[16] Farsightedness can also result particularly if the lens is highly subluxated. Subluxation (partial dislocation) of the lens can be detected clinically in about 60% of people with Marfan syndrome by the use of a slit-lamp biomicroscope.[16] If the lens subluxation is subtle, then imaging with high-resolution ultrasound biomicroscopy might be used.[17]
Other signs and symptoms affecting the eye include increased length along an axis of the globe, myopia, corneal flatness, strabismus, exotropia, and esotropia.[15] Those with MFS are also at a high risk for early glaucoma and early cataracts.[16]
Cardiovascular system
The most serious signs and symptoms associated with Marfan syndrome involve the cardiovascular system: undue fatigue, shortness of breath, heart palpitations, racing heartbeats, or chest pain radiating to the back, shoulder, or arm. Cold arms, hands, and feet can also be linked to MFS because of inadequate circulation. A heart murmur, abnormal reading on an ECG, or symptoms of angina can indicate further investigation. The signs of regurgitation from prolapse of the mitral or aortic valves (which control the flow of blood through the heart) result from cystic medial degeneration of the valves, which is commonly associated with MFS (see mitral valve prolapse, aortic regurgitation). However, the major sign that would lead a doctor to consider an underlying condition is a dilated aorta or an aortic aneurysm. Sometimes, no heart problems are apparent until the weakening of the connective tissue (cystic medial degeneration) in the ascending aorta causes an aortic aneurysm or aortic dissection, a surgical emergency. An aortic dissection is most often fatal and presents with pain radiating down the back, giving a tearing sensation.[18]
Because underlying connective tissue abnormalities cause MFS, the incidence of dehiscence of prosthetic mitral valve is increased.[19] Care should be taken to attempt repair of damaged heart valves rather than replacement.[20]
Lungs
Individuals with Marfan Syndrome may be affected by various lung-related problems. One study found that only 37% of the patient sample studied (mean age 32±14 years; M 45%) had normal lung function.[21] Spontaneous pneumothorax is common.[22] In spontaneous unilateral pneumothorax, air escapes from a lung and occupies the pleural space between the chest wall and a lung. The lung becomes partially compressed or collapsed. This can cause pain, shortness of breath, cyanosis, and, if not treated, death. Other possible pulmonary manifestations of MFS include sleep apnea[23] and idiopathic obstructive lung disease.[24] Pathologic changes in the lungs have been described such as cystic changes, emphysema, pneumonia, bronchiectasis, bullae, apical fibrosis and congenital malformations such as middle lobe hypoplasia.[25]
Nervous system
Dural ectasia, the weakening of the connective tissue of the dural sac encasing the spinal cord, can result in a loss of quality of life. It can be present for a long time without producing any noticeable symptoms. Symptoms that can occur are lower back pain, leg pain, abdominal pain, other neurological symptoms in the lower extremities, or headaches – symptoms which usually diminish when lying flat. On X-ray, however, dural ectasia is not often visible in the early stages. A worsening of symptoms might warrant an MRI of the lower spine. Dural ectasia that has progressed to this stage would appear in an MRI as a dilated pouch wearing away at the lumbar vertebrae.[26] Other spinal issues associated with MFS include degenerative disc disease, spinal cysts, and dysfunction of the autonomic nervous system.
Genetics
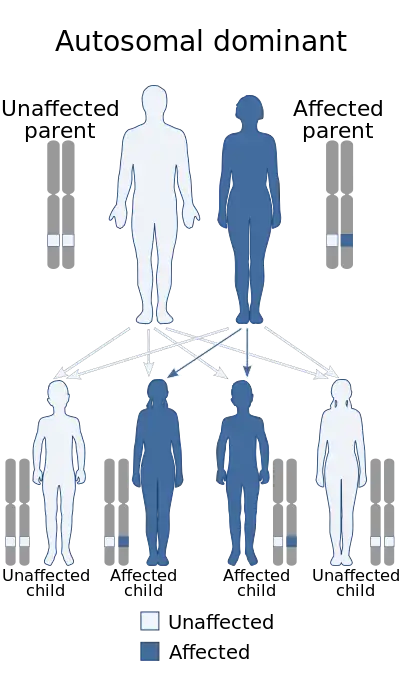
Each parent with the condition has a 50% risk of passing the genetic defect on to any child due to its autosomal dominant nature. Most individuals with MFS have another affected family member. About 75% of cases are inherited.[1] On the other hand, about 15–30% of all cases are due to de novo genetic mutations;[27] such spontaneous mutations occur in about one in 20,000 births. Marfan syndrome is also an example of dominant negative mutation and haploinsufficiency.[28][29] It is associated with variable expressivity; complete penetrance has been definitively documented.[30]
Pathogenesis
Marfan syndrome is caused by mutations in the FBN1 gene on chromosome 15,[31] which encodes fibrillin 1, a glycoprotein component of the extracellular matrix. Fibrillin-1 is essential for the proper formation of the extracellular matrix, including the biogenesis and maintenance of elastic fibers. The extracellular matrix is critical for both the structural integrity of connective tissue, but also serves as a reservoir for growth factors.[27] Elastic fibers are found throughout the body, but are particularly abundant in the aorta, ligaments and the ciliary zonules of the eye; consequently, these areas are among the worst affected.
A transgenic mouse has been created carrying a single copy of a mutant fibrillin-1, a mutation similar to that found in the human gene known to cause MFS. This mouse strain recapitulates many of the features of the human disease and promises to provide insights into the pathogenesis of the disease. Reducing the level of normal fibrillin 1 causes a Marfan-related disease in mice.[32]
Transforming growth factor beta (TGF-β) plays an important role in MFS. Fibrillin-1 directly binds a latent form of TGF-β, keeping it sequestered and unable to exert its biological activity. The simplest model suggests reduced levels of fibrillin-1 allow TGF-β levels to rise due to inadequate sequestration. Although how elevated TGF-β levels are responsible for the specific pathology seen with the disease is not proven, an inflammatory reaction releasing proteases that slowly degrade the elastic fibers and other components of the extracellular matrix is known to occur. The importance of the TGF-β pathway was confirmed with the discovery of the similar Loeys–Dietz syndrome involving the TGFβR2 gene on chromosome 3, a receptor protein of TGF-β.[33] Marfan syndrome has often been confused with Loeys–Dietz syndrome, because of the considerable clinical overlap between the two pathologies.[34]
Marfanoid–progeroid–lipodystrophy syndrome
Marfanoid–progeroid–lipodystrophy syndrome (MPL), also referred to as Marfan lipodystrophy syndrome (MFLS), is a variant of MFS in which Marfan symptoms are accompanied by features usually associated with neonatal progeroid syndrome (also referred to as Wiedemann–Rautenstrauch syndrome) in which the levels of white adipose tissue are reduced.[35] Since 2010, evidence has been accumulating that MPL is caused by mutations near the 3'-terminus of the FBN1 gene.[36][37] It has been shown that these people are also deficient in asprosin, a gluco-regulatory protein hormone which is the C-terminal cleavage product of profibrillin. The levels of asprosin seen in these people were lower than expected for a heterozygous genotype, consistent with a dominant negative effect.[38]
Diagnosis
Diagnostic criteria of MFS were agreed upon internationally in 1996.[39] However, Marfan syndrome is often difficult to diagnose in children, as they typically do not show symptoms until reaching pubescence.[40] A diagnosis is based on family history and a combination of major and minor indicators of the disorder, rare in the general population, that occur in one individual – for example: four skeletal signs with one or more signs in another body system such as ocular and cardiovascular in one individual. The following conditions may result from MFS, but may also occur in people without any known underlying disorder.
- Aortic aneurysm or dilation[41]
- Arachnodactyly
- GERD
- Bicuspid aortic valve
- Cysts
- Cystic medial necrosis
- Degenerative disc disease
- Deviated septum[42]
- Dural ectasia
- Early cataracts
- Early glaucoma[43]
- Early osteoarthritis[44]
- Ectopia lentis
- Emphysema[45]
- Iris coloboma[46]
- Above-average height
- Heart palpitations[47]
- Hernias[48]
- High-arched palate
- Hypermobility of the joints
- Kyphosis (hunched back)
- Leaky heart valve
- Malocclusion
- Micrognathia (small lower jaw)[46]
- Mitral valve prolapse[41]
- Myopia (nearsightedness)
- Obstructive lung disease
- Osteopenia (low bone density)[49]
- Pectus carinatum or excavatum
- Pes planus (flat feet)[50]
- Pneumothorax (collapsed lung)[51]
- Retinal detachment[52]
- Scoliosis[53]
- Sleep apnea[23]
- Stretch marks not from pregnancy[54] or obesity
- Teeth crowded[54]
- "Narrow, thin face"[46]
- Temporomandibular joint dysfunction (TMD)[55]
Revised Ghent nosology
In 2010, the Ghent nosology was revised, and new diagnostic criteria superseded the previous agreement made in 1996. The seven new criteria can lead to a diagnosis:[56][57]
In the absence of a family history of MFS:
- Aortic root Z-score ≥ 2 AND ectopia lentis
- Aortic root Z-score ≥ 2 AND an FBN1 mutation
- Aortic root Z-score ≥ 2 AND a systemic score* > 7 points
- Ectopia lentis AND an FBN1 mutation with known aortic pathology
In the presence of a family history of MFS (as defined above):
- Ectopia lentis
- Systemic score* ≥ 7
- Aortic root Z-score ≥ 2
- Points for systemic score:
- Wrist AND thumb sign = 3 (wrist OR thumb sign = 1)
- Pectus carinatum deformity = 2 (pectus excavatum or chest asymmetry = 1)
- Hindfoot deformity = 2 (plain pes planus = 1)
- Dural ectasia = 2
- Protrusio acetabuli = 2
- pneumothorax = 2
- Reduced upper segment/lower segment ratio AND increased arm/height AND no severe scoliosis = 1
- Scoliosis or thoracolumbar kyphosis = 1
- Reduced elbow extension = 1
- Facial features (3/5) = 1 (dolichocephaly, enophthalmos, downslanting palpebral fissures, malar hypoplasia, retrognathia)
- Skin striae (stretch marks) = 1
- Myopia > 3 diopters = 1
- Mitral valve prolapse = 1
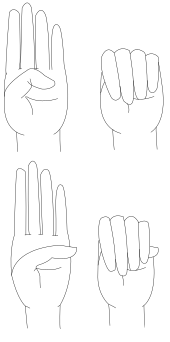
The thumb sign (Steinberg's sign) is elicited by asking the person to flex the thumb as far as possible and then close the fingers over it. A positive thumb sign is where the entire distal phalanx is visible beyond the ulnar border of the hand, caused by a combination of hypermobility of the thumb as well as a thumb which is longer than usual.
The wrist sign (Walker-Murdoch sign) is elicited by asking the person to curl the thumb and fingers of one hand around the other wrist. A positive wrist sign is where the little finger and the thumb overlap, caused by a combination of thin wrists and long fingers.[58]
Differential diagnosis
Many other disorders can produce the same type of body characteristics as Marfan syndrome.[59] Genetic testing and evaluating other signs and symptoms can help to differentiate these. The following are some of the disorders that can manifest as "marfanoid":
Management
There is no cure for Marfan syndrome, but life expectancy has increased significantly over the last few decades and is now similar to that of the average person.[61]
Regular checkups are recommended to monitor the health of the heart valves and the aorta. Marfan syndrome is treated by addressing each issue as it arises and, in particular, preventive medication even for young children to slow progression of aortic dilation. The goal of this treatment strategy is to slow the progression of aortic dilation and prevent any damage to heart valves by eliminating heart arrythmias, minimizing the heart rate, and lowering the person's blood pressure.[62]
Physical activity
The American Heart Association made the following recommendations for people with Marfan syndrome with no or mild aortic dilation:[63][64]
- Probably permissible activities: bowling, golf, skating (but not ice hockey), snorkeling, brisk walking, treadmill, stationary biking, modest hiking, and tennis (doubles and singles).
- Intermediate risk: basketball (both full- and half-court), racquetball, squash, running (sprinting and jogging), skiing (downhill and cross-country), soccer, touch (flag) football, baseball, softball, biking, lap swimming, motorcycling, and horseback riding.
- High risk: bodybuilding, weightlifting (non-free and free weights), ice hockey, rock climbing, windsurfing, surfing, and scuba diving.
Medication
Management often includes the use of beta blockers such as propranolol or if not tolerated calcium channel blockers or ACE inhibitors.[3][4] Beta blockers are used to reduce the stress exerted on the aorta and to decrease aortic dilation.[16]
Surgery
If the dilation of the aorta progresses to a significant-diameter aneurysm, causes a dissection or a rupture, or leads to failure of the aortic or other valve, then surgery (possibly a composite aortic valve graft or valve-sparing aortic root replacement) becomes necessary. Although aortic graft surgery (or any vascular surgery) is a serious undertaking it is generally successful if undertaken on an elective basis.[65] Surgery in the setting of acute aortic dissection or rupture is considerably more problematic. Elective aortic valve/graft surgery is usually considered when aortic root diameter reaches 50 millimeters (2.0 inches), but each case needs to be specifically evaluated by a qualified cardiologist. New valve-sparing surgical techniques are becoming more common.[66] As people with Marfan syndrome live longer, other vascular repairs are becoming more common, e.g., repairs of descending thoracic aortic aneurysms and aneurysms of vessels other than the aorta.
The skeletal and ocular manifestations of Marfan syndrome can also be serious, although not life-threatening. These symptoms are usually treated in an appropriate manner for the condition, such as with pain medications or muscle relaxants. Because Marfan syndrome may cause asymptomatic spinal abnormalities, any spinal surgery contemplated on a person Marfan should only follow detailed imaging and careful surgical planning, regardless of the indication for surgery. The ocular complications of MFS can often be treated with surgery. Ectopia lentis can be treated, as artificial lenses can be surgically implanted. In addition, surgery can address glaucoma and cataracts.[16]
Treatment of a spontaneous pneumothorax is dependent on the volume of air in the pleural space and the natural progression of the individual's condition. A small pneumothorax might resolve without active treatment in one to two weeks. Recurrent pneumothoraces might require chest surgery. Moderately sized pneumothoraces might need chest drain management for several days in a hospital. Large pneumothoraces are likely to be medical emergencies requiring emergency decompression.
As an alternative approach, custom-built supports for the aortic root are also being used.[67] As of 2020 this procedure has been used in over 300 people with the first case occurring in 2004.[68][69]
Pregnancy
During pregnancy, even in the absence of preconception cardiovascular abnormality, women with Marfan syndrome are at significant risk of aortic dissection, which is often fatal even when rapidly treated. Women with Marfan syndrome, then, should receive a thorough assessment prior to conception, and echocardiography should be performed every six to 10 weeks during pregnancy, to assess the aortic root diameter. For most women, safe vaginal delivery is possible.[70]
Prenatal testing can be performed in females with Marfan syndrome to determine if the condition has been inherited in their child.[40] At 10 to 12 weeks of pregnancy, examining a piece of placental tissue through a test called chorionic villus sampling can be performed to make a diagnosis.[40] Another prenatal test can be performed called amniocentesis at 16 to 18 weeks of pregnancy.[40]
Marfan syndrome is expressed dominantly. This means a child with one parent a bearer of the gene has a 50% probability of getting the syndrome. In 1996, the first preimplantation genetic testing (PGT) therapy for Marfan was conducted;[71] in essence PGT means conducting a genetic test on early-stage IVF embryo cells and discarding those embryos affected by the Marfan mutation.
Prognosis
Prior to modern cardiovascular surgical techniques and medications such as losartan, and metoprolol, the prognosis of those with Marfan syndrome was not good: a range of untreatable cardiovascular issues was common. Lifespan was reduced by at least a third, and many died in their teens and twenties due to cardiovascular problems. Today, cardiovascular symptoms of Marfan syndrome are still the most significant issues in diagnosis and management of the disease, but adequate prophylactic monitoring and prophylactic therapy offers something approaching a normal lifespan, and more manifestations of the disease are being discovered as more patients live longer.[72] Women with Marfan syndrome live longer than men.[15]
Epidemiology
Marfan syndrome affects males and females equally,[73] and the mutation shows no ethnic or geographical bias.[8] Estimates indicate about 1 in 5,000 to 10,000 individuals have Marfan syndrome.[3]
History
Marfan syndrome is named after Antoine Marfan,[9] the French pediatrician who first described the condition in 1896 after noticing striking features in a five-year-old girl.[10][74] The gene linked to the disease was first identified by Francesco Ramirez at the Mount Sinai Medical Center in New York City in 1991.[75]
Famous patients
Famous people who have had Marfan syndrome:
- Isaiah Austin[76]
- Javier Botet[77]
- Austin Carlile[78]
- Bradford Cox[79]
- Euell Gibbons[80][81]
- Emmanuel Giroux
- Flo Hyman[82]
- Jonathan Jeanne[83]
- Vincent Schiavelli
- Troye Sivan[84]
- John Tavener[85]
- Peter Mayhew[86]
Other historical figures and celebrities have appeared on lists of people with Marfan syndrome, but from case to case the evidence is speculative, questionable, or even refuted.
- Akhenaten (Artistic depictions show many physical characteristics of people with Marfan, such as elongated skull and larger pelvis with enlarged thighs and spindly calves.[87] Most Egyptologists as of 2021 argue that Akhenaten's portrayals are not the results of a genetic or medical condition, but rather should be interpreted as stylized portrayals influenced by Atenism.[88][89])
- Osama bin Laden (Marfan rumors deemed "likely false" by journalists[90][91])
- Julius Caesar (speculative)
- Robert Johnson (speculative[92])
- Jonathan Larson (speculative)[93]
- Abraham Lincoln (disputed speculative diagnosis—Lincoln's DNA has not been tested[94])
- Niccolò Paganini (speculative[95])
- Michael Phelps (disputed by Phelps[82])
- Sergei Rachmaninoff (disputed: "Rachmaninov did not clearly exhibit any of the other clinical characteristics typical of Marfan's. . . . Nor did he express any of the clinical effects of a Marfan-related syndrome"[96])
- Tutankhamen (did not have Marfan Syndrome[97][98][99])
See also
Bibliography
- Lorenz, Megaera. "Lorenz, Maegara "The Mystery of Akhenaton: Genetics or Aesthetics"". Heptune.com. Archived from the original on February 8, 2010. Retrieved March 21, 2010.
- Montserrat, Dominic (2003) [2000]. Akhenaten: History, Fantasy and Ancient Egypt (1st paperback ed.). London; New York: Routledge. ISBN 0415301866.
- Reeves, Nicholas (2019) [2001]. Akhenaten: Egypt's False Prophet (Electronic ed.). London; New York: Thames & Hudson. ISBN 978-0-500-29469-7. LCCN 00108868.
References
- "What Is Marfan Syndrome?". NHLBI, NIH. October 1, 2010. Archived from the original on 6 May 2016. Retrieved 16 May 2016.
- "How Is Marfan Syndrome Diagnosed?". NHLBI, NIH. October 1, 2010. Archived from the original on 11 June 2016. Retrieved 16 May 2016.
- "Marfan Syndrome". National Organization for Rare Disorders. 2017. Archived from the original on 12 November 2019. Retrieved 5 November 2016.
- "How Is Marfan Syndrome Treated?". NHLBI, NIH. October 1, 2010. Archived from the original on 11 June 2016. Retrieved 16 May 2016.
- Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, et al. (July 2010). "The revised Ghent nosology for the Marfan syndrome". Journal of Medical Genetics. 47 (7): 476–485. doi:10.1136/jmg.2009.072785. hdl:1854/LU-1013955. PMID 20591885. S2CID 13895128. Archived from the original on 2017-08-29. Retrieved 2016-01-10.
- Nistri S, De Cario R, Sticchi E, Spaziani G, Della Monica M, Giglio S, et al. (September 2021). "Differential Diagnosis between Marfan Syndrome and Loeys-Dietz Syndrome Type 4: A Novel Chromosomal Deletion Covering TGFB2". Genes. 12 (10): 1462. doi:10.3390/genes12101462. PMC 8536070. PMID 34680857.
- "What Are the Signs and Symptoms of Marfan Syndrome?". NHLBI, NIH. October 1, 2010. Archived from the original on 11 June 2016. Retrieved 16 May 2016.
- Keane MG, Pyeritz RE (May 2008). "Medical management of Marfan syndrome". Circulation. 117 (21): 2802–2813. doi:10.1161/CIRCULATIONAHA.107.693523. PMID 18506019.
estimated prevalence of 1 case per 3000 to 5000 individuals
- Marfan, Antoine (1896). "Un cas de déformation congénitale des quartre membres, plus prononcée aux extrémitiés, caractérisée par l'allongement des os avec un certain degré d'amincissement [A case of congenital deformation of the four limbs, more pronounced at the extremities, characterized by elongation of the bones with some degree of thinning]". Bulletins et Mémoires de la Société Médicale des Hôpitaux de Paris (in French). 13 (3rd series): 220–226. OCLC 493643386. NAID 10014879958.
- "Antoine Bernard-Jean Marfan". Whonamedit?. Archived from the original on 8 March 2016. Retrieved 16 May 2016.
- "Lifelong drug therapy the key to coping with Marfan syndrome". South China Morning Post. 2 December 2013. Archived from the original on 18 February 2022. Retrieved 18 February 2022.
- Zoabi, A.; Lavie, G. (19 November 2014). "Marfan syndrome and the thumb sign". QJM. 108 (6): 509. doi:10.1093/qjmed/hcu224. PMID 25411342. Archived from the original on 18 February 2022. Retrieved 18 February 2022.
- "Steinberg sign (Marfan disease)". Radiopaedia. Archived from the original on 2021-10-17. Retrieved 2021-10-17.
- Van de Velde S, Fillman R, Yandow S (March 2006). "Protrusio acetabuli in Marfan syndrome. History, diagnosis, and treatment". The Journal of Bone and Joint Surgery. American Volume. 88 (3): 639–646. doi:10.2106/JBJS.E.00567. PMID 16510833.
- "OMIM Entry - # 154700 - MARFAN SYNDROME; MFS". omim.org. Archived from the original on 2019-09-08. Retrieved 2016-08-08.
- "About Marfan Syndrome". Genome.gov. Archived from the original on 2020-03-02. Retrieved 2020-03-02.
- Liu, Yi-Zhi; Liu, Yu-Hua; Wu, Ming-Xing; Luo, Li-Xia; Zhang, Xin-Yu; Cai, Xiao-Yu; Chen, Xiu-Qi (March 2004). "[Clinical applications of ultrasound biomicroscopy in diagnosis and treatment of lens subluxation]". [Zhonghua Yan Ke Za Zhi] Chinese Journal of Ophthalmology. 40 (3): 186–189. ISSN 0412-4081. PMID 15307991. Archived from the original on 2022-10-09. Retrieved 2022-10-09.
- "Aortic dissection - Symptoms and causes". Mayo Clinic. Archived from the original on 2022-10-09. Retrieved 2022-10-09.
- Zipes, Libby Bonow Braunwald (2005). Braunwald's Heart Disease ~ A Textbook of Cardiovascular Medicine, Seventh Edition. United States of America: Elseview Saunders. p. 1894. ISBN 978-0-7216-0509-8.
- Mick, Stephanie L.; Keshavamurthy, Suresh; Gillinov, A. Marc (May 2015). "Mitral valve repair versus replacement". Annals of Cardiothoracic Surgery. 4 (3): 230–237. doi:10.3978/j.issn.2225-319X.2015.03.01. ISSN 2225-319X. PMC 4533076. PMID 26309824.
- Cerveri I, Corsico A (2012). "Pulmonary involvement in patients with Marfan syndrome". European Respiratory Journal. 40: 3124.
- Siepe M, Löffelbein F (June 2009). "[The Marfan syndrome and related connective tissue disorders]". Medizinische Monatsschrift Fur Pharmazeuten. 32 (6): 213–219. PMID 19554831.
- Kohler M, Blair E, Risby P, Nickol AH, Wordsworth P, Forfar C, Stradling JR (February 2009). "The prevalence of obstructive sleep apnoea and its association with aortic dilatation in Marfan's syndrome". Thorax. 64 (2): 162–166. doi:10.1136/thx.2008.102756. PMID 18852161.
- Corsico AG, Grosso A, Tripon B, Albicini F, Gini E, Mazzetta A, et al. (June 2014). "Pulmonary involvement in patients with Marfan Syndrome". Panminerva Medica. 56 (2): 177–182. PMID 24994580. Archived from the original on 2021-11-20. Retrieved 2021-11-20.
- Dyhdalo K, Farver C (December 2011). "Pulmonary histologic changes in Marfan syndrome: a case series and literature review". American Journal of Clinical Pathology. 136 (6): 857–863. doi:10.1309/AJCP79SNDHGKQFIN. PMID 22095370.
- "Marfan Syndrome". Mayo Clinic. Archived from the original on January 10, 2007. Retrieved January 12, 2007.
- Robbins SL, Cotran RS, Robbins SL, Kumar V (1998). Robbins Pathologic Basis of Disease. Philadelphia: W.B Saunders Company. ISBN 978-0-7216-7335-6.
- Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, et al. (July 2004). "Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome". The Journal of Clinical Investigation. 114 (2): 172–181. doi:10.1172/JCI20641. PMC 449744. PMID 15254584.
- Judge DP, Dietz HC (December 2005). "Marfan's syndrome". Lancet. 366 (9501): 1965–1976. doi:10.1016/S0140-6736(05)67789-6. PMC 1513064. PMID 16325700.
- von Kodolitsch, Yskert; Robinson, Peter N (June 2007). "Marfan syndrome: an update of genetics, medical and surgical management". Heart. 93 (6): 755–760. doi:10.1136/hrt.2006.098798. ISSN 1355-6037. PMC 1955191. PMID 17502658.
- McKusick VA (July 1991). "The defect in Marfan syndrome". Nature. 352 (6333): 279–281. Bibcode:1991Natur.352..279M. doi:10.1038/352279a0. PMID 1852198.
- Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, Bunton T, et al. (March 1999). "Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1". Proceedings of the National Academy of Sciences of the United States of America. 96 (7): 3819–3823. Bibcode:1999PNAS...96.3819P. doi:10.1073/pnas.96.7.3819. PMC 22378. PMID 10097121.
- Entrez Gene (2007). "TGFBR2 transforming growth factor, beta receptor II" (Entrez gene entry). NCBI. Archived from the original on January 13, 2007. Retrieved January 11, 2007.
- "Related Disorders: Loeys–Dietz". National Marfan Foundation. Archived from the original on September 25, 2006. Retrieved January 11, 2007.
- "OMIM Entry - #616914 - MARFAN LIPODYSTROPHY SYNDROME; MFLS". omim.org. Archived from the original on 2018-11-30. Retrieved 2016-12-06.
- Graul-Neumann LM, Kienitz T, Robinson PN, Baasanjav S, Karow B, Gillessen-Kaesbach G, et al. (November 2010). "Marfan syndrome with neonatal progeroid syndrome-like lipodystrophy associated with a novel frameshift mutation at the 3' terminus of the FBN1-gene". American Journal of Medical Genetics. Part A. 152A (11): 2749–2755. doi:10.1002/ajmg.a.33690. PMID 20979188. S2CID 26408208.
- Jacquinet A, Verloes A, Callewaert B, Coremans C, Coucke P, de Paepe A, et al. (April 2014). "Neonatal progeroid variant of Marfan syndrome with congenital lipodystrophy results from mutations at the 3' end of FBN1 gene". European Journal of Medical Genetics. 57 (5): 230–234. doi:10.1016/j.ejmg.2014.02.012. PMID 24613577.
- Romere C, Duerrschmid C, Bournat J, Constable P, Jain M, Xia F, et al. (April 2016). "Asprosin, a Fasting-Induced Glucogenic Protein Hormone". Cell. 165 (3): 566–579. doi:10.1016/j.cell.2016.02.063. PMC 4852710. PMID 27087445.
- De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE (April 1996). "Revised diagnostic criteria for the Marfan syndrome". American Journal of Medical Genetics. 62 (4): 417–426. doi:10.1002/(SICI)1096-8628(19960424)62:4<417::AID-AJMG15>3.0.CO;2-R. PMID 8723076.
- "Marfan Syndrome | Testing and Diagnosis | Boston Children's Hospital". www.childrenshospital.org. Archived from the original on 2020-03-02. Retrieved 2020-03-02.
- "Marfan Syndrome". The Lecturio Medical Concept Library. Archived from the original on 23 January 2021. Retrieved 10 August 2021.
- Finkbohner R, Johnston D, Crawford ES, Coselli J, Milewicz DM (February 1995). "Marfan syndrome. Long-term survival and complications after aortic aneurysm repair". Circulation. 91 (3): 728–733. doi:10.1161/01.CIR.91.3.728. PMID 7828300.
- "Marfan Syndrome: Signs and Symptoms". www.ucsfhealth.org. Archived from the original on 2010-06-17. Retrieved 2009-08-28.
- "What is Marfan Syndrome?". Marfan Trust. Archived from the original on 2015-06-10. Retrieved 2015-06-01.
- "Marfan Syndrome: The Similarities to Copper Deficiency". www.ctds.info. Archived from the original on 2009-02-21. Retrieved 2009-08-29.
- MedlinePlus Encyclopedia: Marfan syndrome
- "Marfan syndrome". Genetics Home Reference. U.S. National Institute of Health. Archived from the original on 2009-08-29. Retrieved 2009-08-28.
- Fitzgibbons RJ, Forse RA (February 2015). "Clinical practice. Groin hernias in adults". The New England Journal of Medicine. 372 (8): 756–763. doi:10.1056/NEJMcp1404068. PMID 25693015.
- Kohlmeier L, Gasner C, Bachrach LK, Marcus R (October 1995). "The bone mineral status of patients with Marfan syndrome". Journal of Bone and Mineral Research. 10 (10): 1550–1555. doi:10.1002/jbmr.5650101017. PMID 8686512. S2CID 23492402.
- Northwestern Memorial Center for Heart Valve Disease. Marfan syndrome Archived 2012-04-22 at the Wayback Machine
- "Pneumothorax". The Lecturio Medical Concept Library. Archived from the original on 10 August 2021. Retrieved 10 August 2021.
- "Retinal Detachment". The Lecturio Medical Concept Library. 21 October 2020. Archived from the original on 10 August 2021. Retrieved 10 August 2021.
- "Scoliosis". The Lecturio Medical Concept Library. Archived from the original on 10 August 2021. Retrieved 10 August 2021.
- "About Marfan Syndrome: Features". National Marfan Foundation. Archived from the original on 2009-08-20. Retrieved 2009-08-28.
- "Living with Marfan Syndrome: Dental issues". National Marfan Foundation. Archived from the original on 2009-09-06. Retrieved 2009-08-28.
- "2010 Revised Ghent Nosology". National Marfan Foundation. Archived from the original on 2011-01-14. Retrieved 2011-01-31.
- Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, et al. (July 2010). "The revised Ghent nosology for the Marfan syndrome" (PDF). Journal of Medical Genetics. 47 (7): 476–485. doi:10.1136/jmg.2009.072785. hdl:1854/LU-1013955. OCLC 857424767. PMID 20591885. S2CID 13895128. Archived (PDF) from the original on 10 January 2016.
- Julia A. McMillan, Ralph D. Feigin, Catherine DeAngelis, M. Douglas Jones. Oski's Pediatrics: Principles & Practice. Lippincott Williams & Wilkins, 2006
- Rimoin DL, Connor JM, Pyeritz RE, et al. (2007). Emery and RImoin's Principles and Practice of Medical Genetics. 5th ed. Philadelphia, Pennsylvania: Churchill Livingstone Elsevier.
- Greally & GeneReviews 2010
- "Questions and Answers about Marfan Syndrome". Niams.nih.gov. Archived from the original on 9 April 2014. Retrieved 23 June 2014.
- Foundation, The Marfan (2013-09-02). "Common Blood Pressure Drug Reduces Aortic Enlargement in Marfan Syndrome". Marfan Foundation. Archived from the original on 2022-10-09. Retrieved 2022-10-09.
- Maron BJ, Chaitman BR, Ackerman MJ, Bayés de Luna A, Corrado D, Crosson JE, et al. (June 2004). "Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases". Circulation. 109 (22): 2807–2816. doi:10.1161/01.cir.0000128363.85581.e1. OCLC 110943757. PMID 15184297.
- "Marfan Syndrome and How Sports Science is Saving Lives". Archived from the original on 2022-08-26. Retrieved 2022-08-26.
- "Elective Aortic Root Surgery in Marfan Syndrome Appears Safe and Durable: Presented at STS" (Press release). Doctor's Guide. January 31, 2008. Archived from the original on November 20, 2008. Retrieved January 13, 2009.
See also:- Cameron DE, Vricella LA (2005). "Valve-sparing aortic root replacement in Marfan syndrome". Seminars in Thoracic and Cardiovascular Surgery. Pediatric Cardiac Surgery Annual. 8 (1): 103–111. doi:10.1053/j.pcsu.2005.03.001. PMID 15818365.
- Gott VL, Cameron DE, Alejo DE, Greene PS, Shake JG, Caparrelli DJ, Dietz HC (February 2002). "Aortic root replacement in 271 Marfan patients: a 24-year experience". The Annals of Thoracic Surgery. 73 (2): 438–443. doi:10.1016/S0003-4975(01)03336-7. PMID 11845856.
- Bethea BT, Fitton TP, Alejo DE, Barreiro CJ, Cattaneo SM, Dietz HC, et al. (September 2004). "Results of aortic valve-sparing operations: experience with remodeling and reimplantation procedures in 65 patients". The Annals of Thoracic Surgery. 78 (3): 767–72, discussion 767–72. doi:10.1016/j.athoracsur.2004.03.040. PMID 15336989.
- "Heart Surgery for Marfan Syndrome". Mayo Clinic. Archived from the original on December 18, 2006. Retrieved January 12, 2007.
- Treasure T, Petrou M, Rosendahl U, Austin C, Rega F, Pirk J, Pepper J (September 2016). "Personalized external aortic root support: a review of the current status". European Journal of Cardio-Thoracic Surgery. 50 (3): 400–404. doi:10.1093/ejcts/ezw078. PMID 27032474.
- Treasure T, Golesworthy T, Pepper J (September 2017). "Practical clinical applications of 3-D printing in cardiovascular surgery". Journal of Thoracic Disease. 9 (9): 2792–2797. doi:10.21037/jtd.2017.08.63. PMC 5708385. PMID 29221242.
- Nemec P, Pepper J, Fila P (September 2020). "Personalized external aortic root support". Interactive Cardiovascular and Thoracic Surgery. 31 (3): 342–345. doi:10.1093/icvts/ivaa111. PMID 32761056.
- Haskett D, Doyle JJ, Gard C, Chen H, Ball C, Estabrook MA, et al. (January 2012). "Altered tissue behavior of a non-aneurysmal descending thoracic aorta in the mouse model of Marfan syndrome". Cell and Tissue Research. 347 (1): 267–277. doi:10.1007/s00441-011-1270-y. PMID 22105919. S2CID 14333291. Archived from the original on July 6, 2009. Retrieved June 25, 2007.
- Harton GL, Tsipouras P, Sisson ME, Starr KM, Mahoney BS, Fugger EF, et al. (September 1996). "Preimplantation genetic testing for Marfan syndrome". Molecular Human Reproduction. 2 (9): 713–715. doi:10.1093/molehr/2.9.713. PMID 9239687.
- Keane MG, Pyeritz RE (May 2008). "Medical management of Marfan syndrome". Circulation. 117 (21): 2802–2813. doi:10.1161/CIRCULATIONAHA.107.693523. PMID 18506019.
- Fusar-Poli P, Klersy C, Stramesi F, Callegari A, Arbustini E, Politi P (May 2008). "Determinants of quality of life in Marfan syndrome". Psychosomatics. 49 (3): 243–248. doi:10.1176/appi.psy.49.3.243. PMID 18448780.
- Johns Hopkins Comprehensive Marfan Center. Archived 2008-10-15 at the Wayback Machine Johns Hopkins Medicine. Retrieved on January 6, 2009.
- Brown P (July 27, 1991). "Marfan syndrome linked to gene". Archived 2015-01-29 at the Wayback Machine New Scientist. Retrieved on August 11, 2008.
- "Isaiah Austin: Doctors have cleared my return". December 2016. Archived from the original on 2023-01-23. Retrieved 2021-11-19.
- Cooper, Kelly-Leigh (May 27, 2019). "Javier Botet: Meet the actor behind Hollywood's monsters". BBC News. Archived from the original on November 27, 2021. Retrieved November 27, 2021.
- Trendell, Andrew (December 1, 2016). "Of Mice and Men's Austin Carlile's devastating message to fans: 'I won't get better'". NME. Archived from the original on November 27, 2021. Retrieved November 27, 2021.
- "As His Recent Bizarre Behaviour Shows, Deerhunter's Bradford Cox Hasn't Mellowed – and That's Why We Need Him". NME. 9 December 2015. Archived from the original on 19 November 2021. Retrieved 19 November 2021.
- "Remember Euell Gibbons, the man who would eat a pine tree on TV?". Metv.com. 2021-08-13. Archived from the original on 2022-08-16. Retrieved 2022-06-27.
- "Euell Gibbons – Wild Food Adventures". Wildfoodadventures.com. 30 September 2015. Archived from the original on 2022-03-19. Retrieved 2022-06-27.
- "Is it a genetic flaw that makes Phelps the greatest?". 16 August 2008. Archived from the original on 19 November 2021. Retrieved 19 November 2021.
- "NBA prospect Jeanne diagnosed with Marfan syndrome". Sports Illustrated. Archived from the original on 2021-11-19. Retrieved 2021-11-19.
- "Troye Sivan fires back at body-shaming Twitter post". PopBuzz. Archived from the original on 2022-01-13. Retrieved 2022-01-13.
- "Health | Sir John Tavener". Archived from the original on 2021-11-19. Retrieved 2021-11-19.
- "In Good Company". Marfan Trust. Archived from the original on 2023-08-20. Retrieved 2023-08-20.
- Lorenz 2010.
- Reeves 2019, pp. 154–155.
- Montserrat 2003.
- "Osama bin Laden Health Rumors: Fact or Fiction?". ABC News. Archived from the original on 2021-11-17. Retrieved 2021-11-19.
- "New life for an old rumor: Was bin Laden 'Marfanoid'?". NBC News. 11 May 2011. Archived from the original on 19 November 2021. Retrieved 19 November 2021.
- Connell D (2 September 2006). "Retrospective blues: Robert Johnson—an open letter to Eric Clapton". BMJ. 333 (7566): 489. doi:10.1136/bmj.333.7566.489. PMC 1557967.
- "Tick, Tick… Boom! On Netflix Continues the Legacy of Jonathan Larson". 11 November 2021. Archived from the original on 19 November 2021. Retrieved 19 November 2021.
- "Did Abraham Lincoln Have Marfan Syndrome? – Clinical Correlations". Archived from the original on 2021-11-19. Retrieved 2021-11-19.
- Wolf P (November 2001). "Creativity and chronic disease. Niccolo Paganini (1782-1840)". The Western Journal of Medicine. 175 (5): 345. doi:10.1136/ewjm.175.5.345. PMC 1071620. PMID 11694491.
- Ramachandran M, Aronson JK (October 2006). "The diagnosis of art: Rachmaninov's hand span". Journal of the Royal Society of Medicine. 99 (10): 529–530. doi:10.1177/014107680609901015. PMC 1592053. PMID 17066567.
- Hawass Z, Gad YZ, Ismail S, Khairat R, Fathalla D, Hasan N, et al. (February 2010). "Ancestry and pathology in King Tutankhamun's family". JAMA. 303 (7): 638–647. doi:10.1001/jama.2010.121. PMID 20159872.
- "Egypt's King Tut born of incestuous marriage: Tests". Reuters. 17 February 2010. Archived from the original on 19 November 2021. Retrieved 19 November 2021.
- "How Did King Tut Die?". 10 August 2023. Archived from the original on 19 November 2021. Retrieved 19 November 2021.