List of primary immunodeficiencies
This is a list of primary immunodeficiencies (PID), which are immune deficiencies that are not secondary to another condition.
The International Union of Immunological Societies recognizes nine classes of primary immunodeficiencies, totaling approximately 430 conditions.[1][2] A 2014 update of the classification guide added a 9th category and added 30 new gene defects from the prior 2009 version.[3][4] The most recent classification was released in 2019.[5] The number of identified conditions continues to grow over time as more research is done.
The impact of primary immunodeficiencies ranges from mild to severe based on the condition.[6]
Combined T and B–cell immunodeficiencies
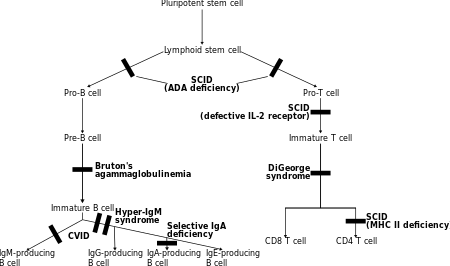
In these disorders both T lymphocytes and often B lymphocytes, regulators of adaptive immunity, are dysfunctional or decreased in number. The main members are various types of severe combined immunodeficiency (SCID).[7]
- T-/B+ SCID (T cells predominantly absent):
- γc deficiency
- JAK3 deficiency
- Interleukin-7 receptor-α deficiency
- CD45 deficiency
- CD3δ, CD3ε, or CD3ζ deficiency
- Coronin-1A deficiency
- LAT (gene) deficiency
- T-/B- SCID (both T and B cells absent)
- RAG 1/2 deficiency
- DCLRE1C (Artemis) deficiency
- XLF (protein)/Cernunnos deficiency
- DNA PKcs deficiency
- DNA ligase type IV deficiency
- adenosine deaminase (ADA) deficiency
- reticular dysgenesis
- Omenn syndrome
- CD40 ligand deficiency
- CD40 deficiency
- CD3γ deficiency
- CD8 deficiency
- ICOS deficiency
- ZAP70 deficiency
- Ca++ channel deficiency
- MHC class I deficiency (with mutations in TAP1, TAP2, TAPBP, or B2M)
- MHC class II deficiency (with mutations in CIITA, RFXANK, RFX5, or RFXAP)
- CD25 deficiency
- CD27 deficiency
- STAT5b deficiency
- ITK deficiency
- SH2D1A deficiency (XLP1)
- MAGT1 deficiency
- DOCK2 deficiency
- DOCK8 deficiency
- RhoH deficiency
- Activated PI3K delta syndrome
- MALT1 deficiency
- BCL10 deficiency
- BCL11B deficiency
- CARD11 deficiency
- MST1 deficiency
- TCRα deficiency
- LCK deficiency
- IL-21 deficiency
- IL-21R deficiency
- UNC119 deficiency
- NIK deficiency
- OX40 deficiency
- IKBKB deficiency
- TFRC deficiency
- Moesin deficiency
- RELB deficiency
- Cartilage hair hypoplasia
- LRBA deficiency
Predominantly antibody deficiencies
In primary antibody deficiencies, one or more isotypes of immunoglobulin are decreased or don't function properly. These proteins, generated by plasma cells, normally bind to pathogens, targeting them for destruction.[7]
- Absent B cells with a resultant severe reduction of all types of antibody: X-linked agammaglobulinemia (btk deficiency, or Bruton's agammaglobulinemia), μ-Heavy chain deficiency, l 5 deficiency, Igα deficiency, BLNK deficiency, thymoma with immunodeficiency
- B cells low but present or normal, but with reduction in 2 or more isotypes (usually IgG & IgA, sometimes IgM): common variable immunodeficiency (CVID), CD19 deficiency, TACI (TNFRSF13B) deficiency, BAFF receptor deficiency.
- Normal numbers of B cells with decreased IgG and IgA and increased IgM: Hyper-IgM syndromes
- Normal numbers of B cells with isotype or light chain deficiencies: heavy chain deletions, kappa chain deficiency, isolated IgG subclass deficiency, IgA with IgG subclass deficiency, selective immunoglobulin A deficiency
- Specific antibody deficiency to specific antigens with normal B cell and normal Ig concentrations
- Transient hypogammaglobulinemia of infancy (THI)
Other well defined immunodeficiency syndrome
A number of syndromes escape formal classification but are otherwise recognizable by particular clinical or immunological features.[7]
- Immunodeficiency with thrombocytopenia
- Wiskott–Aldrich syndrome
- WIP deficiency
- ARPC1B deficiency
- DNA repair defects not causing isolated SCID:
- Ataxia-telangiectasia
- Ataxia-like syndrome
- Nijmegen breakage syndrome
- Bloom syndrome
- Immunodeficiency–centromeric instability–facial anomalies syndrome (ICF1, 2, 3, and 4)
- PMS2 deficiency
- RIDDLE syndrome (RNF168 deficiency)
- MCM4 deficiency
- FILS syndrome (POLE deficiency)
- POLE2 deficiency
- LIG1 deficiency
- NSMCE3 deficiency
- Hebo deficiency
- GINS1 deficiency
- DiGeorge syndrome (when associated with thymic defects)
- TBX1 deficiency
- CHARGE syndrome (CHD7 deficiency or SEMA3E deficiency)
- Winged helix/FOXN1 deficiency
- Chromosome 10p13-p14 deletion
- Immuno-osseous dysplasias (abnormal development of the skeleton with immune problems):
- Cartilage–hair hypoplasia
- Schimke syndrome
- MYSM1 deficiency
- MOPD1 deficiency
- EXTL3 deficiency
- Hyper IgE syndromes
- Job syndrome (STAT3 deficiency)
- Comel-Netherton syndrome
- PGM3 deficiency
- Hypohidrotic ectodermal dysplasia
- Calcium channel defects
- ORAI1 deficiency
- STIM1 deficiency
- Transcobalamin 2 deficiency
- Immunodeficiency with multiple intestinal atresias (TTC7A deficiency)
- Hepatic venoocclusive disease with immunodeficiency (VODI)
- Vici syndrome
- Purine nucleoside phosphorylase (PNP) deficiency
- AR-DKC (autosomal dominant dyskeratosis congenital)
- Hermansky–Pudlak syndrome type 2
- Chronic mucocutaneous candidiasis
- HOIL1 deficiency
- HOIP deficiency
- XL-dyskeratosis congenita (Hoyeraal-Hreidarsson syndrome)
- Hennekam lymphangiectasia-lymphedema syndrome
- Kabuki syndrome
- MTHFD1 deficiency
- STAT5b deficiency
- IKAROS deficiency
Diseases of immune dysregulation
In certain conditions, the regulation rather than the intrinsic activity of parts of the immune system is the predominant problem.[7]
- Immunodeficiency with hypopigmentation or albinism: Chédiak–Higashi syndrome, Griscelli syndrome type 2
- Familial hemophagocytic lymphohistiocytosis: perforin deficiency, UNC13D deficiency, syntaxin 11 deficiency
- X-linked lymphoproliferative syndrome
- Syndromes with autoimmunity:
- (a) Autoimmune lymphoproliferative syndrome: type 1a (CD95 defects), type 1b (Fas ligand defects), type 2a (CASP10 defects), type 2b (CASP8 defects)
- (b) APECED (autoimmune polyendocrinopathy with candidiasis and ectodermal dystrophy)
- (c) IPEX (immunodysregulation polyendocrinopathy enteropathy X-linked syndrome)
- (d) CD25 deficiency
Congenital defects of phagocyte number, function, or both
Phagocytes are the cells that engulf and ingest pathogens (phagocytosis), and destroy them with chemicals. Monocytes/macrophages as well as granulocytes are capable of this process. In certain conditions, either the number of phagocytes is reduced or their functional capacity is impaired.[7]
- Severe Congenital Neutropenia: due to ELA2 deficiency (with myelodysplasia)
- Severe Congenital Neutropenia: due to GFI1 deficiency (with T/B lymphopenia)
- Elastase deficiency
- Kostmann syndrome (HAX1 deficiency)
- Neutropenia with cardiac and urogenital malformations
- Glycogen storage disease type 1b
- Cohen syndrome
- Clericuzio syndrome
- Cyclic neutropenia
- X-linked neutropenia/myelodysplasia
- P14 deficiency
- HYOU1 deficiency
- JAGN1 deficiency
- SMARCD2 deficiency
- 3-Methylglutaconic aciduria
- Leukocyte adhesion deficiency type 1
- Leukocyte adhesion deficiency type 2
- Leukocyte adhesion deficiency type 3
- RAC2 deficiency (Neutrophil immunodeficiency syndrome)
- Beta-actin deficiency
- G-CSF-receptor deficiency
- Localized juvenile periodontitis
- Papillon–Lefèvre syndrome
- Specific granule deficiency
- Shwachman–Diamond syndrome
- WDR1 deficiency
- Cystic fibrosis
- Chronic granulomatous disease: X-linked or autosomal (CYBA, NCF1, NCF2, NCF4)
- IL-12 and IL-23 β1 chain deficiency
- IL-12p40 deficiency
- Glucose-6-phosphate dehydrogenase deficiency class 1
- Interferon γ receptor 1 deficiency
- Interferon γ receptor 2 deficiency
- STAT1 deficiency
- MKL1 deficiency
- AD hyper-IgE
- AR hyper-IgE
- Pulmonary alveolar proteinosis
- MonoMac syndrome (GATA2 deficiency)
Defects in innate immunity
Several rare conditions are due to defects in the innate immune system, which is a basic line of defense that is independent of the more advanced lymphocyte-related systems. Many of these conditions are associated with skin problems.[7]
- Interleukin 12 receptor, beta 1 deficiency
- IL-12p40 deficiency
- Interferon gamma receptor 1 deficiency
- Interferon gamma receptor 2 deficiency
- Tyk2 deficiency
- JAK1 loss-of-function
- ISG15 deficiency
- RORc deficiency
- STAT1 deficiency, gain-of-function mutation
- STAT2 deficiency
- IRF7 deficiency
- CD16 deficiency
- IRF8 deficiency
- IFNAR2 deficiency
- TLR pathway deficiencies
- MDA5 deficiency
- Epidermodysplasia verruciformis
- WHIM syndrome (warts, hypogammaglobulinaemia, infections, myelokathexis)
- EVER1 and EVER2 deficiency
- Herpes simplex encephalitis
- CARD9 deficiency
- Chronic mucocutaneous candidiasis
- Trypanosomiasis
- RPSA deficiency with congenital asplenia
- HMOX deficiency with congenital asplenia
- CLCN7 deficiency with osteoporosis
- OSTM1 deficiency with osteoporosis
- Hidradenitis suppurativa
Autoinflammatory disorders
Rather than predisposing for infections, most of the autoinflammatory disorders lead to excessive inflammation. Many manifest themselves as periodic fever syndromes. They may involve various organs directly, as well as predisposing for long-term damage (e.g. by leading to amyloid deposition).[7]
- Familial Mediterranean fever
- Aicardi–Goutières syndrome with TREX1, SAMHD1 or IFIH1 mutations
- Spondyloenchondro-dysplasia with immune dysregulation (ACP5 mutation)
- STING-associated vasculopathy with onset in infancy
- X-linked reticulate pigmentary disorder
- USP18 deficiency
- CANDLE (Chronic atypical neutrophilic dermatitis with lipodystrophy)
- Singleton-Merten syndrome
- TNF receptor associated periodic syndrome (TRAPS)
- Hyper-IgD syndrome (Mevalonate kinase deficiency)
- CIAS1-related diseases:
- NLRP1 deficiency
- PAPA syndrome (pyogenic sterile arthritis, pyoderma gangrenosum, acne)
- ADAM17 deficiency
- Blau syndrome
- Majeed syndrome (Chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anemia)
- DIRA (deficiency of the IL-1 receptor antagonist)
- DITRA (deficiency of IL-36 receptor antagonist)
- CARD14 mediated psoriasis (CAMPS)
- Cherubism
- COPA defect
- Otulipenia/ORAS
Complement deficiencies
The complement system is part of the innate as well as the adaptive immune system; it is a group of circulating proteins that can bind pathogens and form a membrane attack complex. Complement deficiencies are the result of a lack of any of these proteins. They may predispose to infections but also to autoimmune conditions.[7]
- C1q deficiency (lupus-like syndrome, rheumatoid disease, infections)
- C1r deficiency (idem)
- C1s deficiency
- C4 deficiency (lupus-like syndrome)
- C2 deficiency (lupus-like syndrome, vasculitis, polymyositis, pyogenic infections)
- C3 deficiency (recurrent pyogenic infections)
- C5 deficiency (Neisserial infections, SLE)
- C6 deficiency (idem)
- C7 deficiency (idem, vasculitis)
- C8a deficiency
- C8b deficiency
- C9 deficiency (Neisserial infections)
- C1-inhibitor deficiency (hereditary angioedema)
- Factor I deficiency (pyogenic infections)
- Factor H deficiency (haemolytic-uraemic syndrome, membranoproliferative glomerulonephritis)
- Factor D deficiency (Neisserial infections)
- Properdin deficiency (Neisserial infections)
- MBP deficiency (pyogenic infections)
- MASP2 deficiency
- Complement receptor 3 deficiency
- Membrane cofactor protein (CD46) deficiency
- Membrane attack complex inhibitor (CD59) deficiency
- Paroxysmal nocturnal hemoglobinuria
- Ficolin 3 deficiency
- Properdin deficiency
- Factor I deficiency
- Factor H deficiency
- Thrombomodulin deficiency
- CHAPEL disease
Phenocopies of primary immune deficiencies
References
- Bousfiha, Aziz; Jeddane, Leïla; Picard, Capucine; Ailal, Fatima; Bobby Gaspar, H.; Al-Herz, Waleed; Chatila, Talal; Crow, Yanick J. (2018). "The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies". Journal of Clinical Immunology. 38 (1): 129–143. doi:10.1007/s10875-017-0465-8. ISSN 0271-9142. PMC 5742599. PMID 29226301.
- Tangye, Stuart G.; Al-Herz, Waleed; Bousfiha, Aziz; Chatila, Talal; Cunningham-Rundles, Charlotte; Etzioni, Amos; Franco, Jose Luis; Holland, Steven M.; Klein, Christoph; Morio, Tomohiro; Ochs, Hans D. (2020-01-01). "Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee". Journal of Clinical Immunology. 40 (1): 24–64. doi:10.1007/s10875-019-00737-x. ISSN 1573-2592. PMC 7082301. PMID 31953710.
- Waleed Al-Herz; Aziz Bousfiha; Jean-Laurent Casanova; et al. (2014). "Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency" (PDF). Frontiers in Immunology. 5 (162): 1–33. doi:10.3389/fimmu.2014.00162. PMC 4001072. PMID 24795713.
- Notarangelo L, Casanova JL, Conley ME, et al. (2006). "Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee Meeting in Budapest, 2005". J. Allergy Clin. Immunol. 117 (4): 883–96. doi:10.1016/j.jaci.2005.12.1347. PMID 16680902.
- Tangye, Stuart G.; Al-Herz, Waleed; Bousfiha, Aziz; Chatila, Talal; Cunningham-Rundles, Charlotte; Etzioni, Amos; Franco, Jose Luis; Holland, Steven M.; Klein, Christoph; Morio, Tomohiro; Ochs, Hans D. (2020-01-01). "Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee". Journal of Clinical Immunology. 40 (1): 24–64. doi:10.1007/s10875-019-00737-x. ISSN 1573-2592. PMC 7082301. PMID 31953710.
- "Common Variable Immune Deficiency". NORD (National Organization for Rare Disorders). Retrieved 5 March 2019.
- Notarangelo LD, Fischer A, Geha RS, et al. (December 2009). "Primary immunodeficiencies: 2009 update: The International Union of Immunological Societies (IUIS) Primary Immunodeficiencies (PID) Expert Committee". J. Allergy Clin. Immunol. 124 (6): 1161–78. doi:10.1016/j.jaci.2009.10.013. PMC 2797319. PMID 20004777.