Ribose-phosphate diphosphokinase
Ribose-phosphate diphosphokinase (or phosphoribosyl pyrophosphate synthetase or ribose-phosphate pyrophosphokinase) is an enzyme that converts ribose 5-phosphate into phosphoribosyl pyrophosphate (PRPP).[1][2] It is classified under EC 2.7.6.1.
| Ribose-phosphate diphosphokinase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
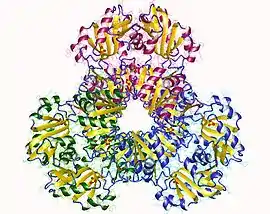 Phosphoribosyl pyrophosphate synthase 1, hexamer, Human | |||||||||
| Identifiers | |||||||||
| EC no. | 2.7.6.1 | ||||||||
| CAS no. | 9031-46-3 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
| phosphoribosyl pyrophosphate synthetase 1 | |||||||
|---|---|---|---|---|---|---|---|
| Identifiers | |||||||
| Symbol | PRPS1 | ||||||
| NCBI gene | 5631 | ||||||
| HGNC | 9462 | ||||||
| OMIM | 311850 | ||||||
| RefSeq | NM_002764 | ||||||
| UniProt | P60891 | ||||||
| Other data | |||||||
| EC number | 2.7.6.1 | ||||||
| Locus | Chr. X q21-q27 | ||||||
| |||||||
| phosphoribosyl pyrophosphate synthetase 2 | |||||||
|---|---|---|---|---|---|---|---|
| Identifiers | |||||||
| Symbol | PRPS2 | ||||||
| NCBI gene | 5634 | ||||||
| HGNC | 9465 | ||||||
| OMIM | 311860 | ||||||
| RefSeq | NM_002765 | ||||||
| UniProt | P11908 | ||||||
| Other data | |||||||
| EC number | 2.7.6.1 | ||||||
| Locus | Chr. X pter-q21 | ||||||
| |||||||
The enzyme is involved in the synthesis of nucleotides (purines and pyrimidines), cofactors NAD and NADP, and amino acids histidine and tryptophan,[1][2][3] linking these biosynthetic processes to the pentose phosphate pathway, from which the substrate ribose 5-phosphate is derived. Ribose 5-phosphate is produced by the HMP Shunt Pathway from Glucose-6-Phosphate. The product phosphoribosyl pyrophosphate acts as an essential component of the purine salvage pathway and the de novo synthesis of purines. Dysfunction of the enzyme would thereby undermine purine metabolism. Ribose-phosphate pyrophosphokinase exists in bacteria, plants, and animals, and there are three isoforms of human ribose-phosphate pyrophosphokinase.[2] In humans, the genes encoding the enzyme are located on the X chromosome.[2]
Reaction mechanism

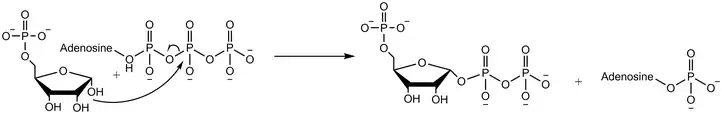
Ribose-phosphate diphosphokinase transfers the diphosphoryl group from Mg-ATP (Mg2+ coordinated to ATP) to ribose 5-phosphate.[2] The enzymatic reaction begins with the binding of ribose 5-phosphate, followed by binding of Mg-ATP to the enzyme. In the transition state upon binding of both substrates, the diphosphate is transferred. The enzyme first releases AMP before releasing the product phosphoribosyl pyrophosphate.[4] Experiments using oxygen 18 labelled water demonstrate that the reaction mechanism proceeds with the nucleophilic attack of the anomeric hydroxyl group of ribose 5-phosphate on the beta-phosphorus of ATP in an SN2 reaction.[5]
Structure
.png.webp)
.png.webp)
Crystallization and X-ray diffraction studies elucidated the structure of the enzyme, which was isolated by cloning, protein expression, and purification techniques. One subunit of ribose-phosphate diphosphokinase consists of 318 amino acids; the active enzyme complex consists of three homodimers (or six subunits, a hexamer). The structure of one subunit is a five-stranded parallel beta sheet (the central core) surrounded by four alpha helices at the N-terminal domain and five alpha helices at the C-terminal domain, with two short anti-parallel beta-sheets extending from the core.[2] The catalytic site of the enzyme binds ATP and ribose 5-phosphate. The flexible loop (Phe92–Ser108), pyrophosphate binding loop (Asp171–Gly174), and flag region (Val30–Ile44 from an adjacent subunit) comprise the ATP binding site, located at the interface between two domains of one subunit. The flexible loop is so named because of its large variability in conformation.[6] The ribose 5-phosphate binding site consists of residues Asp220–Thr228, located in the C-terminal domain of one subunit.[2][6] The allosteric site, which binds ADP, consists of amino acid residues from three subunits.[2]
Function
The product of this reaction, phosphoribosyl pyrophosphate (PRPP), is used in numerous biosynthesis (de novo and salvage) pathways. PRPP provides the ribose sugar in de novo synthesis of purines and pyrimidines, used in the nucleotide bases that form RNA and DNA. PRPP reacts with orotate to form orotidylate, which can be converted to uridylate (UMP). UMP can then be converted to the nucleotide cytidine triphosphate (CTP). The reaction of PRPP, glutamine, and ammonia forms 5-Phosphoribosyl-1-amine, a precursor to inosinate (IMP), which can ultimately be converted to adenosine triphosphate (ATP) or guanosine triphosphate (GTP). PRPP plays a role in purine salvage pathways by reacting with free purine bases to form adenylate, guanylate, and inosinate.[7][8] PRPP is also used in the synthesis of NAD: the reaction of PRPP with nicotinic acid yields the intermediate nicotinic acid mononucleotide.[9]
Regulation
Ribose-phosphate diphosphokinase requires Mg2+ for activity; the enzyme acts only on ATP coordinated with Mg2+. Ribose-phosphate diphosphokinase is regulated by phosphorylation and allostery. It is activated by phosphate and inhibited by ADP; it is suggested that phosphate and ADP compete for the same regulatory site. At normal concentrations, phosphate activates the enzyme by binding to its allosteric regulatory site. However, at high concentrations, phosphate is shown to have an inhibitory effect by competing with the substrate ribose 5-phosphate for binding at the active site. ADP is the key allosteric inhibitor of ribose-phosphate diphosphokinase. It has been shown that at lower concentrations of the substrate ribose 5-phosphate, ADP may inhibit the enzyme competitively. Ribose-phosphate pyrophosphokinase is also inhibited by some of its downstream biosynthetic products.[2][6]
Role in disease
Because its product is a key compound in many biosynthetic pathways, ribose-phosphate diphosphokinase is involved in some rare disorders and X-linked recessive diseases. Mutations that lead to super-activity (increased enzyme activity or de-regulation of the enzyme) result in purine and uric acid overproduction. Super-activity symptoms include gout, sensorineural hearing loss,[10] weak muscle tone (hypotonia), impaired muscle coordination (ataxia), hereditary peripheral neuropathy,[11] and neurodevelopmental disorder.[12][13][14] Mutations that lead to loss-of-function in ribose-phosphate diphosphokinase result in Charcot-Marie-Tooth disease and Arts syndrome.[15]
References
- Visentin LP, Hasnain S, Gallin W (July 1977). "Ribosomal protein S1/S1A in bacteria". FEBS Lett. 79 (2): 258–63. doi:10.1016/0014-5793(77)80799-0. PMID 330231. S2CID 38926849.
- Li S, Lu Y, Peng B, Ding J (January 2007). "Crystal structure of human phosphoribosylpyrophosphate synthetase 1 reveals a novel allosteric site". Biochem. J. 401 (1): 39–47. doi:10.1042/BJ20061066. PMC 1698673. PMID 16939420.
- Tang W, Li X, Zhu Z, Tong S, Li X, Zhang X, Teng M, Niu L (May 2006). "Expression, purification, crystallization and preliminary X-ray diffraction analysis of human phosphoribosyl pyrophosphate synthetase 1 (PRS1)". Acta Crystallographica Section F. 62 (Pt 5): 432–4. doi:10.1107/S1744309106009067. PMC 2219982. PMID 16682768.
- Fox IH, Kelley WN (April 1972). "Human phosphoribosylpyrophosphate synthetase. Kinetic mechanism and end product inhibition". J. Biol. Chem. 247 (7): 2126–31. doi:10.1016/S0021-9258(19)45500-2. PMID 4335863.
- Miller GA, Rosenzweig S, Switzer RL (December 1975). "Oxygen-18 studies of the mechanism of pyrophosphoryl group transfer catalyzed by phosphoribosylpyrophosphate synthetase". Arch. Biochem. Biophys. 171 (2): 732–6. doi:10.1016/0003-9861(75)90086-7. PMID 173242.
- Eriksen TA, Kadziola A, Bentsen AK, Harlow KW, Larsen S (April 2000). "Structural basis for the function of Bacillus subtilis phosphoribosyl-pyrophosphate synthetase". Nat. Struct. Biol. 7 (4): 303–8. doi:10.1038/74069. PMID 10742175. S2CID 23115108.
- Fox IH, Kelley WN (March 1971). "Phosphoribosylpyrophosphate in man: biochemical and clinical significance". Ann. Intern. Med. 74 (3): 424–33. doi:10.7326/0003-4819-74-3-424. PMID 4324023.
- Jeremy M. Berg; John L. Tymoczko; Lubert Stryer; Gregory J. Gatto Jr. (2012). Biochemistry (7th ed.). New York: W.H. Freeman. ISBN 978-1429229364.
- Rongvaux A, Andris F, Van Gool F, Leo O (July 2003). "Reconstructing eukaryotic NAD metabolism". BioEssays. 25 (7): 683–90. doi:10.1002/bies.10297. PMID 12815723.
- Liu X, Han D, Li J, Han B, Ouyang X, Cheng J, Li X, Jin Z, Wang Y, Bitner-Glindzicz M, Kong X, Xu H, Kantardzhieva A, Eavey RD, Seidman CE, Seidman JG, Du LL, Chen ZY, Dai P, Teng M, Yan D, Yuan H (January 2010). "Loss-of-function mutations in the PRPS1 gene cause a type of nonsyndromic X-linked sensorineural deafness, DFN2". Am. J. Hum. Genet. 86 (1): 65–71. doi:10.1016/j.ajhg.2009.11.015. PMC 2801751. PMID 20021999.
- Kim HJ, Sohn KM, Shy ME, Krajewski KM, Hwang M, Park JH, Jang SY, Won HH, Choi BO, Hong SH, Kim BJ, Suh YL, Ki CS, Lee SY, Kim SH, Kim JW (September 2007). "Mutations in PRPS1, which encodes the phosphoribosyl pyrophosphate synthetase enzyme critical for nucleotide biosynthesis, cause hereditary peripheral neuropathy with hearing loss and optic neuropathy (cmtx5)". Am. J. Hum. Genet. 81 (3): 552–8. doi:10.1086/519529. PMC 1950833. PMID 17701900.
- Becker MA, Smith PR, Taylor W, Mustafi R, Switzer RL (November 1995). "The genetic and functional basis of purine nucleotide feedback-resistant phosphoribosylpyrophosphate synthetase superactivity". J. Clin. Invest. 96 (5): 2133–41. doi:10.1172/JCI118267. PMC 185862. PMID 7593598.
- Zoref E, De Vries A, Sperling O (November 1975). "Mutant feedback-resistant phosphoribosylpyrophosphate synthetase associated with purine overproduction and gout. Phosphoribosylpyrophosphate and purine metabolism in cultured fibroblasts". J. Clin. Invest. 56 (5): 1093–9. doi:10.1172/JCI108183. PMC 301970. PMID 171280.
- "Phosphoribosylpyrophosphate synthetase superactivity". Lister Hill National Center for Biomedical Communications. Retrieved 25 February 2014.
- Synofzik M, Müller Vom Hagen J, Haack TB, Wilhelm C, Lindig T, Beck-Wödl S, Nabuurs SB, van Kuilenburg AB, de Brouwer AP, Schöls L (2014). "X-linked Charcot-Marie-Tooth disease, Arts syndrome, and prelingual non-syndromic deafness form a disease continuum: evidence from a family with a novel PRPS1 mutation". Orphanet J Rare Dis. 9 (1): 24. doi:10.1186/1750-1172-9-24. PMC 3931488. PMID 24528855.
External links
- Uniprot - Ribose-phosphate pyrophosphokinase 1
- GeneReviews/NIH/NCBI/UW entry on Charcot-Marie-Tooth Neuropathy X Type 5
- OMIM entries on Charcot-Marie-Tooth Neuropathy X Type 5
- GeneReviews/NCBI/NIH/UW entry on Arts Syndrome
- GeneReviews/NIH/NCBI/UW entry on Phosphoribosylpyrophosphate Synthetase (PRS) Superactivity
- GeneReviews/NCBI/NIH/UW entry on DFNX1 Nonsyndromic Hearing Loss and Deafness
- Phosphoribosyl+Pyrophosphate+Synthetase at the U.S. National Library of Medicine Medical Subject Headings (MeSH)