Congenital red–green color blindness
Congenital red–green color blindness is an inherited condition that is the root cause of the majority of cases of color blindness. It has no significant symptoms aside from its minor to moderate effect on color vision.[1] It is caused by variation in the functionality of the red and/or green opsin proteins, which are the photosensitive pigment in the cone cells of the retina, which mediate color vision.[1] Males are more likely to inherit red–green color blindness than females, because the genes for the relevant opsins are on the X chromosome.[1] Screening for congenital red–green color blindness is typically performed with the Ishihara or similar color vision test.[1] There is no cure for color blindness.[1]
| Congenital red–green color blindness | |
|---|---|
| Other names | Daltonism; red–green color vision deficiency |
 | |
| An example Ishihara test that may be used to detect red–green color blindness. Those with normal color vision would be able to see a green "74" on an orange background, while those with red–green color blindness would see the green and red hues as much more similar, if not nearly identical to each other. | |
| Specialty | Ophthalmology |
| Symptoms | red–green color blindness |
| Usual onset | congenital |
| Duration | permanent |
| Causes | Genetic (inherited usually X-linked)[1] |
| Diagnostic method | Color vision tests[1] |
| Differential diagnosis | Acquired red–green color blindness |
| Treatment | none |
| Medication | none |
| Frequency | 2-9% males; <1% females |
This form of colorblindness is sometimes referred to historically as daltonism after John Dalton, who had congenital red–green color blindness and was the first to scientifically study it. In other languages, daltonism is still used to describe red–green color blindness, but may also refer colloquially to color blindness in general.
Symptoms
The only significant symptom of congenital red–green color blindness is deficient color vision (color blindness or discromatopsia). Additionally, approximately __% of colorblindness is caused by congenital red–green colorblindness, so the condition and symptom are often difficult to untangle. A red–green color blind subject will have decreased (or no) color discrimination along the red–green axis. This commonly includes the following colors of confusion:
- Cyan and Grey
- Rose-Pink and Grey
- Blue and Purple
- Yellow and Neon Green
- Red, Green, Orange, Brown
- Black and Red (protans)


Classification
| Dimensionality | |||
| Dichromacy | Anomalous Trichromacy | ||
| Cone | L-cone | Protanopia | Protanomaly |
| M-cone | Deuteranopia | Deuteranomaly | |
Congenital red–green color blindness is classified into 1 of 4 groups:
- Protanopia
- Protanomaly
- Deuteranopia
- Deuteranomaly
Each of these groups comprises a prefix and a suffix. The prefix indicates the cone (photopsin) that is affected, with lexemes from Greek, "first" (prot-) or "second" (deuter-) referring to the L- and M-opsins respectively. The suffix indicates the dimensionality of color vision:
- Dichromacy gives the suffix -anopia (from the Greek for "no sight")
- Anomalous trichromacy gives the suffix anomaly (from the Greek for "irregular").
Dimensionality

The dimensionality of normal color vision is trichromatic. This references that a visual system with three distinct cone classes and therefore a three dimensional gamut. Dichromatic color vision only has two distinct cone classes and therefore a two dimensional gamut. With red–green dichromacy, it is the dimension representing the red–green opponent channel that is lost. Anomalous trichromacy is also trichromatic, but the spectral sensitivity of at least one of the cone cells is altered, leading to a gamut that is a different size or shape. In the case of congenital red–green color blindness, the dynamic range of the red–green dimension is decreased when compared to normal color vision.
The dimensionality of the defect is related to the strength/severity, but it is usually much easier clinically to define the severity empirically as mild, moderate and strong (or severe). Anomalous trichromacy can vary in severity from indistinguishable from normal color vision (mild) to indistinguishable from dichromacy (strong). Therefore, the differential diagnosis between anomalous trichromacy and dichromacy is difficult.[2] An example clinical diagnosis would be strong deutan, which could correspond to either deuteranomaly or deuteranopia.
Protan vs. deutan
The two types of congenital red–green color blindness as based on the affected cone are:
- Protan: (2% of males): lacking, or possessing anomalous L-opsins for long-wavelength-sensitive cone cells.
- Deutan: (6% of males): lacking, or possessing anomalous M-opsins for medium-wavelength-sensitive cone cells.
Despite often being called red-blind and green-blind respectively, protan and deutan varieties have very similar phenotypes (color vision), especially when compared to tritan color blindness. The condition is not called red–green color blindness because red and green are indicative colors of confusion, nor because the "red" and "green" cones are affected, but because the red–green opponent process channel is affected. In dichromacy, that channel is equally deactivated regardless of which cone (LWS or MWS) is missing. In anomalous trichromacy, that channel is equally affected regardless of which cone effectively moves towards the other.

The starkest difference is the scoterythrous effect, where reds appear dimmer to protans. This is why protans often confuse red with black, while deutans do not. The protan luminous efficiency function is narrower at long wavelengths, which causes the reds to be darker. This is due to the red cones (which normally cover the red side of the spectrum) either shifting to shorter wavelengths or being missing.
The two are difficult to differentiate with color vision tests, but is most reliably performed with an Anomaloscope. This device measures the proportion of red and green light that must be mixed to perceptually match a yellow reference. Protans add more red than color normals, and deutans add more green.
Mechanism
Genes
The mechanism of congenital red–green color blindness relates to the functionality of cone cells, specifically to the expression of photopsins, the photopigments that 'catch' photons and thereby convert light into chemical signals. A typical human has three distinct photopsins: S-, M- and L-opsins expressed by distinct genes, respectively OPN1SW, OPN1MW or OPN1LW. OPN1MW and OPN1LW are located in a gene cluster (along with a locus control region gene) at position Xq28, at the end of the q arm of the X chromosome in a tandem array.[4] OPN1SW is unrelated to the condition and located on a different chromosome. The genes in the cluster are summarized in the following table:
| Type | OMIM | Gene | Locus | Purpose |
| Locus Control Region | 300824 | LCR[5] | Xq28 | Acts as a promoter of the expression of the two opsin genes thereafter,[5] and ensures that only one of the two opsins (LWS or MWS) is expressed exclusively in each cone.[6] |
| LWS opsin | 300822 | OPN1LW | Xq28 | Encodes the LWS (red) photopsin protein. |
| MWS opsin | 300821 | OPN1MW | Xq28 | Encodes the MWS (green) photopsin protein. |
Differentiating from a duplication event 30-40 MYA,[7] the two opsins are highly homologous (very similar), having only 19 dimorphic sites (amino acids that differ),[8] and are therefore 96% similar.[9] In comparison, either of these opsin genes are only 40% homologous to OPN1SW (encoding the SWS photopsin and located on chromosome 7) or "RHO" (encoding rhodopsin, and located on chromosome 3).[9]
While the two genes share 19 dimorphic sites (amino acids that differ), only 7 of them lead to a functional difference between the genes, i.e. tune the opsin's spectral sensitivity.[8] These 7 functionally dimorphic sites will tune the opsin to a higher (redshift) or lower (blueshift) wavelength. The typical (most common) allele for the OPN1MW gene is blueshifted at every one of these dimorphic sites. Likewise, the typical allele for the OPN1LW gene is redshifted at every one of these dimorphic sites. In other words, the most common alleles of each gene that contribute to normal color vision are as far as part as they can be in the spectrum (about 30 nm) without novel point mutations.
Homologous recombination

During meiosis, homologous recombination between chromosomes of the same type may occur where they exchange a portion of their genes. The exchanged portions are generally equivalent (have the same genes) and this process is called equal homologous recombination.[6] Unequal homologous recombination occurs when the exchanged portions of the chromosomes are not equal, i.e. they don't break in the same spot. This recombination happens often at this locus because the OPN1LW and OPN1MW genes are adjacent and 96% similar.
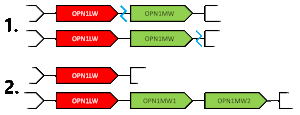
When unequal recombination happens with breaks between the genes (depicted by blue lines), a gene can be essentially deleted from one of the chromosomes. This gene deletion leads to protanopia or deuteranopia (congenital red–green dichromacy).
When unequal recombination happens with breaks in the middle of a gene (e.g. between exons), chimeric genes can be created that contain portions of each of the OPN1LW/OPN1MW genes.
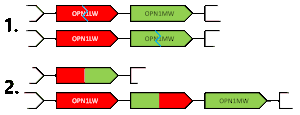
Chimeric gene
A chimeric gene contains exons contributed from the typical alleles of each of the OPN1MW and OPN1LW genes. Due to the similarity between the genes, these chimeras are always functional, but experience a spectral tuning, i.e. a change to the spectral sensitivity. A chimera's spectral sensitivity will lie somewhere between the typical allele peaks (530~560 nm). These chimeric alleles are indicated with an asterisk, either M* or L*. Whether a chimeric gene is described as M* or L* is not based on whether they are closer in the spectrum to the typical M or L allele, but rather is the opposite of the other expressed gene. This means the same chimeric gene could be labeled M* or L* depending on what other genes are in the gene cluster. An individual with protanomaly will have M and L* opsins and an individual with deuteranomaly will have L and M* opsins.
The following table includes the 7 dimorphic sites that contribute to spectral tuning, including their exon and the spectral shift between the typical OPN1MW and OPN1LW exons (as a whole):[6]
| Amino Acid Position | Amino Acid in typical M-opsin | Amino Acid in typical L-opsin | Exon | Spectral Shift |
|---|---|---|---|---|
| 309 | Phenylalanine | Tyrosine | 5 | ±21 nm |
| 285 | Alanine | Threonine | ||
| 277 | Phenylalanine | Tyrosine | ||
| 233 | Serine | Alanine | 4 | ±4 nm |
| 230 | Threonine | Isoleucine | ||
| 180 | Alanine | Serine | 3 | ±3 nm |
| 116 | Tyrosine | Serine | 2 | ±2 nm |
Gene duplications
Gene duplications are one result of the unequal homologous recombination. Either the OPN1LW or OPN1MW can be duplicated, though it is much more commonly the latter. Only 5% of X chromosomes contain multiple OPN1LW genes, but 55% contain multiple OPN1MW genes, sometimes as many as 4.[7] The duplicate genes are sometimes referred to with numerical suffixes, where the OPN1MW gene in the second position is called OPN1MW2. The duplicated genes are always in sequence and can consist of different alleles of the gene, but only the first gene of a duplicate series is ever expressed.
Blue-cone monochromacy
While blue-cone monochromacy exhibits much stronger symptoms than the congenital red–green color blindness (including total color blindness), it follows a very similar mechanism. In most cases, unequal homologous combination must first occur to generate a genotype with a single L/M-opsin gene. Then that gene must experience a nonsense mutation to entirely deactivate it.
Genetics
Congenital means that the condition is present from birth, but is usually used to represent the genetic, inherited basis of the condition. This is in contrast to acquired color blindness that is not present at birth and may be caused by aging, accidents, medication, etc.[10]
Heredity
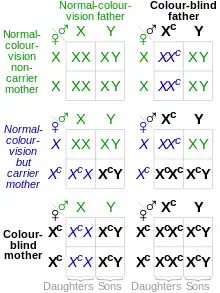
Since the affected opsin genes (OPN1LW and OPN1MW) are on the X chromosome, they are sex-linked, and therefore affect males and females disproportionately. Because the colorblind alleles are recessive, colorblindness follows X-linked recessive inheritance. Males have only one X chromosome (karyotype XY), and females have two (karyotype XX); Because the male only has one allele of each gene, if it is missing or chimeric, the male will be color blind. Because a female has two alleles of each gene (one on each chromosome), if only one allele is mutated, the dominant normal alleles will "override" the mutated, recessive allele and the female will have normal color vision. However, if the female has two mutated alleles, she will still be colorblind. This is why there is a disproportionate prevalence of colorblindness, with ~8% of males exhibiting colorblindness and ~0.5% of females (0.08² = 0.0064 = 0.64%).
Some conclusions from the table include:
- A male cannot inherit colorblindness from his father.
- A colorblind female must have a colorblind father.
- A female must inherit colorblindness alleles from both parents to be colorblind.
- Colorblind females can only produce colorblind males.
- Because carrier females often have a colorblind father, colorblind males often will have a colorblind maternal grandfather (or great-grandfather). In this way, colorblindness is often said to 'skip a generation'.
The Punnett square and this section assume each chromosome only has one affected gene. It also assumes females with two affected chromosomes are affected in the same way.
Genotypes
| Genotype | Result |
|---|---|
| XML Y | Unaffected male |
| XM*L Y | Deutan male |
| XML* Y | Protan male |
| XM*L* Y | Male with possible BCM |
| XML XML | Unaffected female |
| XML XML* XML XM*L |
Female Carrier (possible tetrachromat) |
| XML XM*L* XM*L XML* |
Female Carrier (possible pentachromat) |
| XML* XML* XM*L XM*L |
Protan/Deutan Female |
The table to the right shows the possible allele/chromosome combinations and how their interactions will manifest in an individual. The exact phenotype of some of the combinations depend on whether the affected gene represents an anomalous allele or is missing. For example, the XM*L*Y male may have blue-cone monochromacy if the genes are both missing/non-functional, or near-normal color vision if both genes are anomalous.
- Y : male-only chromosome (no effect on colorblindness)
- X : X chromosome will have two subscripts indicating the alleles present:
- M : normal M opsin allele
- L : normal L opsin allele
- M*: chimeric (or missing) M opsin allele
- L*: chimeric (or missing) L opsin allele
Tetrachromacy in carriers of CVD
Females that are heterozygous for anomalous trichromacy (i.e. carriers) may be tetrachromats.[6] These females have two alleles for either the OPN1MW or OPN1LW gene, and therefore express both the normal and anomalous opsins. Because one X chromosome is inactivated at random in each photoreceptor cell during a female's development, those normal and anomalous opsins will be segregated into their own cone cells, and because these cells have different spectral sensitivity, they can functionally operate as different cone classes. This theoretical female would therefore have cones with peak sensitivities at 420 nm (S cone), 530 nm (M cone), 560 nm (L cone) and the fourth (anomalous) cone between 530 nm and 560 nm (either M* or L* cone).[11][12][13]
If a female is heterozygous for both protanomaly and deuteranomaly, she could be pentachromatic. The degree to which women who are carriers of either protanomaly or deuteranomaly are demonstrably tetrachromatic and require a mixture of four spectral lights to match an arbitrary light is very variable. Jameson et al.[14] have shown that with appropriate and sufficiently sensitive equipment it can be demonstrated that any female carrier of red–green color blindness (i.e. heterozygous protanomaly, or heterozygous deuteranomaly) is a tetrachromat to a greater or lesser extent.
Since the incidence of anomalous trichromacy in males is ~6%, which should equal the incidence of anomalous M opsin or L opsin alleles, it follows that the prevalence of unaffected female carriers of colorblindness (and therefore of potential tetrachromats) is 11.3% (i.e. 94% × 6% × 2), based on the Hardy–Weinberg principle.[15] One such woman has been widely reported to be a true or functional tetrachromat, as she can discriminate colors most other people can't.[12][13]
Diagnosis
Color vision test

The diagnosis of congenital red–green color blindness is usually inferred through psychophysical testing. These color vision tests test detect the color vision phenotype, and not the subject genotype, so are unable to differentiate acquired from congenital red–green color blindness. However, color vision and genotype are highly correlated, especially when acquired color blindness is ruled out.[16] The Ishihara color test is the test most often used to detect red–green deficiencies and most often recognized by the public.[17]
Electroretinography
When psychophysical testing is undesired, an electroretinogram (ERG) can be used instead. An ERG measures the electrical response from the retina as a function of wavelength of light. Due to the shape of spectral sensitivity of cone cells, the peak wavelengths of cone sensitivity can be assumed from an ERG. The peak wavelengths are highly correlated to genotype.[16]
Genetic testing
The genotype can be directly evaluated by sequencing the OPN1MW and OPN1LW genes. The correlation between genotype and phenotype (color vision) are well known, so genetic testing can be a useful supplement to psychophysical color vision tests that may provide incomplete information.[18]
Treatment
Despite much recent improvement in Gene therapy for color blindness, there is currently no FDA approved treatment for congenital red–green color blindness, and otherwise no cure for exists. Management of the condition through the use of color blind glasses to alleviate symptoms or smartphone apps to aid with daily tasks is possible.
Epidemiology
| Dichromacy | 2.1% |
|---|---|
| Deuteranopia | 1.0% |
| Protanopia | 1.1% |
| Anomalous trichromacy | 6.1% |
| Protanomaly | 1.2% |
| Deuteranomaly | 4.9% |
Congenital red–green color blindness affects a large number of individuals, especially individuals of European ancestry, where 8% of men and 0.4% of women exhibit congenital red–green color deficiency.[19] The lower prevalence in females is related to the x-linked inheritance of congenital red–green color blindness, as explained above. Interestingly, even Dalton's very first paper already arrived upon this 8% number:[20]
...it is remarkable that, out of 25 pupils I once had, to whom I explained this subject, 2 were found to agree with me...
— John Dalton, Extraordinary facts relating to the vision of colours: with observations (1798)
Other ethnicities will generally have a lower prevalence of congenital red–green color blindness. The following table summarizes a number of studies performed in different regions.
| Population | Number studied |
% |
|---|---|---|
| Arabs (Druzes) | 337 | 10.0 |
| Aboriginal Australians | 4,455 | 1.9 |
| Belgians | 9,540 | 7.4 |
| Bosnians | 4,836 | 6.2 |
| Britons | 16,180 | 6.6 |
| Chinese | 1,164 | 6.9 |
| DR Congolese | 929 | 1.7 |
| Dutch | 3,168 | 8.0 |
| Fijians | 608 | 0.8 |
| French | 1,243 | 8.6 |
| Germans | 7,861 | 7.7 |
| Hutu | 1,000 | 2.9 |
| Indians (Andhra Pradesh) | 292 | 7.5 |
| Inuit | 297 | 2.5 |
| Iranians | 16,180 | 6.6 |
| Japanese | 259,000 | 4.0 |
| Mexicans | 571 | 2.3 |
| Navajo | 571 | 2.3 |
| Norwegians | 9,047 | 9.0 |
| Russians | 1,343 | 9.2 |
| Scots | 463 | 7.8 |
| Swiss | 2,000 | 8.0 |
| Tibetans | 241 | 5.0 |
| Tswana | 407 | 2.0 |
| Tutsi | 1,000 | 2.5 |
| Serbs | 4,750 | 7.4 |
References
- "Facts About Color Blindness". NEI. February 2015. Archived from the original on 28 July 2016. Retrieved 29 July 2016.
- Simunovic MP (May 2010). "Colour vision deficiency". Eye. 24 (5): 747–55. doi:10.1038/eye.2009.251. PMID 19927164.
- Judd, Deane B. (1979). Contributions to Color Science. Washington D.C. 20234: NBS. p. 316.
{{cite book}}: CS1 maint: location (link) - Alpern M, Lee GB, Maaseidvaag F, Miller SS (January 1971). "Colour vision in blue-cone 'monochromacy'". J. Physiol. 212 (1): 211–33. doi:10.1113/jphysiol.1971.sp009318. PMC 1395698. PMID 5313219.
- Nathans, J; Davenport, C M; Maumenee, I H; Lewis, R A; Hejtmancik, J F; Litt, M; Lovrien, E; Weleber, R; Bachynski, B; Zwas, F; Klingaman, R; Fishman, G (1989). "Molecular genetics of human blue cone monochromacy". Science. 245 (4920): 831–838. Bibcode:1989Sci...245..831N. doi:10.1126/science.2788922. PMID 2788922. S2CID 13093786.
- Neitz, J; Neitz, M (2011). "The genetics of normal and defective color vision". Vision Res. 51 (7): 633–651. doi:10.1016/j.visres.2010.12.002. PMC 3075382. PMID 21167193.
- Davidoff, Candice (2015). Dissertation: Cone opsin gene variants in color blindness and other vision disorders. University of Washington.
- Neitz, Maureen (1 May 2000). "Molecular Genetics of Color Vision and Color Vision Defects". Archives of Ophthalmology. 118 (5): 691–700. doi:10.1001/archopht.118.5.691. PMID 10815162.
- Gardner, Jessica C.; Michaelides, Michel; Holder, Graham E.; Kanuga, Naheed; Webb, Tom R.; Mollon, John D.; Moore, Anthony T.; Hardcastle, Alison J. (1 May 2009). "Blue cone monochromacy: Causative mutations and associated phenotypes". Molecular Vision. 15: 876–884. ISSN 1090-0535. PMC 2676201. PMID 19421413.
- "Acquired colour vision defects". colourblindawareness.org. Archived from the original on 2014-12-16.
- Roth M (13 September 2006). "Some women may see 100,000,000 colors, thanks to their genes". Pittsburgh Post-Gazette. Archived from the original on 8 November 2006.
- Didymus, JohnThomas (Jun 19, 2012), "Scientists find woman who sees 99 million more colors than others", Digital Journal, archived from the original on 2016-02-08
- Jordan G, Deeb SS, Bosten JM, Mollon JD (July 2010). "The dimensionality of color vision in carriers of anomalous trichromacy". Journal of Vision. 10 (8): 12. doi:10.1167/10.8.12. PMID 20884587.
- Jameson KA, Highnote SM, Wasserman LM (June 2001). "Richer color experience in observers with multiple photopigment opsin genes". Psychonomic Bulletin & Review. 8 (2): 244–61. doi:10.3758/BF03196159. PMID 11495112. S2CID 2389566.
- Harrison G, Tanner J, Pilbeam D, Baker P (1988). Human Biology. Oxford: Oxford University Press. pp. 183–187, 287–290. ISBN 978-0-19-854144-8.
- Reference, Genetics Home. "Color vision deficiency". Genetics Home Reference. Retrieved 2019-05-06.
- Gordon N (March 1998). "Colour blindness". Public Health. 112 (2): 81–4. doi:10.1038/sj.ph.1900446. PMID 9581449.
- Davidoff, Candice; Neitz, Maureen; Neitz, Jay (6 September 2016). "Genetic Testing as a New Standard for Clinical Diagnosis of Color Vision Deficiencies". Translational Vision Science & Technology. 5 (5): 2. doi:10.1167/tvst.5.5.2. PMC 5017313. PMID 27622081.
- Birch, Jennifer (1 March 2012). "Worldwide prevalence of red–green color deficiency". Journal of the Optical Society of America A. 29 (3): 313–320. Bibcode:2012JOSAA..29..313B. doi:10.1364/JOSAA.29.000313. PMID 22472762.
- Dalton, John (1798). "Extraordinary Facts relating to the Vision of Colours: With Observations". Manchester Literary and Philosophical Society. Memoirs. England, Manchester. 5 (1): 28–45.
- Harrison, G.A. et al. (1977): Human Biology, Oxford University Press, Oxford, ISBN 0-19-857164-X.
Further reading
- Kaiser PK, Boynton RM (1996). Human color vision. Washington, DC: Optical Society of America. ISBN 978-1-55752-461-4. OCLC 472932250.
- McIntyre D (2002). Colour blindness: causes and effects. Chester: Dalton Publishing. ISBN 978-0-9541886-0-3. OCLC 49204679.
- Dalton J (1798). "Extraordinary facts relating to the vision of colours: with observations". Memoirs of the Literary and Philosophical Society of Manchester. 5: 28–45. OCLC 9879327.
Color topics | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Color science |
|  | ||||||||
| Color philosophy |
| |||||||||
| Color terms |
| |||||||||
| Color organizations | ||||||||||
| Lists | ||||||||||
| Shades of: | ||||||||||
| Related | ||||||||||
| ||||||||||