Antigenic variation
Antigenic variation or antigenic alteration refers to the mechanism by which an infectious agent such as a protozoan, bacterium or virus alters the proteins or carbohydrates on its surface and thus avoids a host immune response, making it one of the mechanisms of antigenic escape. It is related to phase variation. Antigenic variation not only enables the pathogen to avoid the immune response in its current host, but also allows re-infection of previously infected hosts. Immunity to re-infection is based on recognition of the antigens carried by the pathogen, which are "remembered" by the acquired immune response. If the pathogen's dominant antigen can be altered, the pathogen can then evade the host's acquired immune system. Antigenic variation can occur by altering a variety of surface molecules including proteins and carbohydrates. Antigenic variation can result from gene conversion,[1] site-specific DNA inversions,[2] hypermutation,[3] or recombination of sequence cassettes.[4] The result is that even a clonal population of pathogens expresses a heterogeneous phenotype.[5] Many of the proteins known to show antigenic or phase variation are related to virulence.[6]
In bacteria
Antigenic variation in bacteria is best demonstrated by species of the genus Neisseria (most notably, Neisseria meningitidis and Neisseria gonorrhoeae, the gonococcus); species of the genus Streptococcus and the Mycoplasma. The Neisseria species vary their pili (protein polymers made up of subunits called pilin which play a critical role in bacterial adhesion, and stimulate a vigorous host immune response) and the Streptococci vary their M-protein.
In the bacterium Borrelia burgdorferi, the cause of Lyme disease, the surface lipoprotein VlsE can undergo recombination which results in antigenic diversity. The bacterium carries a plasmid that contains fifteen silent vls cassettes and one functional copy of vlsE. Segments of the silent cassettes recombine with the vlsE gene, generating variants of the surface lipoprotein antigen.[7]
In protozoa
Antigenic variation is employed by a number of different protozoan parasites. Trypanosoma brucei and Plasmodium falciparum are some of the best studied examples.
Trypanosoma brucei
Trypanosoma brucei, the organism that causes sleeping sickness,
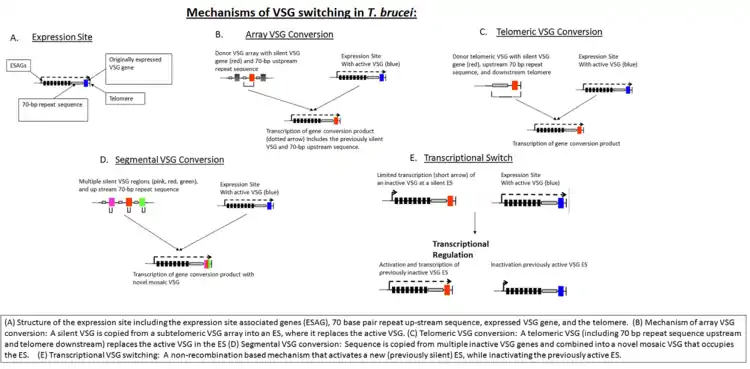
replicates extracellularly in the bloodstream of infected mammals and is subjected to numerous host defense mechanisms including the complement system, and the innate and adaptive immune systems. To protect itself, the parasite decorates itself with a dense, homogeneous coat (~10^7 molecules) of the variant surface glycoprotein (VSG).
In the early stages of invasion, the VSG coat is sufficient to protect the parasite from immune detection. The host eventually identifies the VSG as a foreign antigen and mounts an attack against the microbe. However, the parasite's genome has over 1,000 genes that code for different variants of the VSG protein, located on the subtelomeric portion of large chromosomes, or on intermediate chromosomes. These VSG genes become activated by gene conversion in a hierarchical order: telomeric VSGs are activated first, followed by array VSGs, and finally pseudogene VSGs.[8] Only one VSG is expressed at any given time. Each new gene is switched in turn into a VSG expression site (ES).[9] This process is partially dependent on homologous recombination of DNA, which is mediated in part by the interaction of the T. brucei BRCA2 gene with RAD51 (however, this is not the only possible mechanism, as BRCA2 variants still display some VSG switching).[9]
In addition to homologous recombination, transcriptional regulation is also important in antigen switching, since T. brucei has multiple potential expression sites. A new VSG can either be selected by transcriptional activation of a previously silent ES, or by recombination of a VSG sequence into the active ES (see figure, "Mechanisms of VSG Switching in T. brucei").[8] Although the biological triggers that result in VSG switching are not fully known, mathematical modeling suggests that the ordered appearance of different VSG variants is controlled by at least two key parasite-derived factors: differential activation rates of parasite VSG and density-dependent parasite differentiation.[10][11]
Plasmodium falciparum
Plasmodium falciparum, the major etiologic agent of human malaria, has a very complex life cycle that occurs in both humans and mosquitoes. While in the human host, the parasite spends most of its life cycle within hepatic cells and erythrocytes (in contrast to T. brucei which remains extracellular). As a result of its mainly intracellular niche, parasitized host cells which display parasite proteins must be modified to prevent destruction by the host immune defenses. In the case of Plasmodium, this is accomplished via the dual purpose Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1). PfEMP1 is encoded by the diverse family of genes known as the var family of genes (approximately 60 genes in all). The diversity of the gene family is further increased via a number of different mechanisms including exchange of genetic information at telomeric loci, as well as meiotic recombination. The PfEMP1 protein serves to sequester infected erythrocytes from splenic destruction via adhesion to the endothelium. Moreover, the parasite is able to evade host defense mechanisms by changing which var allele is used to code the PfEMP1 protein.[12] Like T. brucei, each parasite expresses multiple copies of one identical protein. However, unlike T. brucei, the mechanism by which var switching occurs in P. falciparum is thought to be purely transcriptional.[13] Var switching has been shown to take place soon after invasion of an erythrocyte by a P. falciparum parasite.[14] Fluorescent in situ hybridization analysis has shown that activation of var alleles is linked to altered positioning of the genetic material to distinct "transcriptionally permissive" areas.[15]
In viruses
Different virus families have different levels of ability to alter their genomes and trick the immune system into not recognizing. Some viruses have relatively unchanging genomes like paramyxoviruses while others like influenza have rapidly changing genomes that inhibit our ability to create long lasting vaccines against the disease. Viruses in general have much faster rate of mutation of their genomes than human or bacterial cells. In general viruses with shorter genomes have faster rates of mutation than longer genomes since they have a faster rate of replication.[16] It was classically thought that viruses with an RNA genome always had a faster rate of antigenic variation than those with a DNA genome because RNA polymerase lacks a mechanism for checking for mistakes in translation but recent work by Duffy et al. shows that some DNA viruses have the same high rates of antigenic variation as their RNA counterparts.[16] Antigenic variation within viruses can be categorized into 6 different categories called antigenic drift, shift, rift, lift, sift, and gift
Antigenic drift: point mutations that occur through imperfect replication of the viral genome. All viruses exhibit genetic drift over time but the amount that they are able to drift without incurring a negative impact on their fitness varies between families.
Antigenic shift: reassortment of the viral genome that occurs when a single host cell is co-infected with two unique virus particles. As the viruses replicate, they reassort and the genes of the two species get mixed up when packaged into a new budding virus. For influenza, this process could yield up to 256 new variations of the virus, and meaningful antigenic shift events tend to occur every couple of decades.
Antigenic rift: Recombination of viral gene. This occurs when there are again two viral cells that infect the same host cell. In this instance the viruses recombine with pieces of each gene creating a new gene instead of simply switching out genes. Recombination has been extensively studied in avian influenza strains as to how the genetics of H5N1 have changed over time.[17]
Antigenic sift: direct transmission with a zoonotic strain of a virus. This occurs when a human is infected during a spillover event.
Antigenic lift: Viral transmission of host derived gene. Some viruses steal host genes and then incorporate them into their own viral genome, encoding genes that sometimes give them an increased virulence. An example of this is the pox virus vaccinia which encoded a viral growth factor that is very similar to the human growth factor and thought to be stolen from the human genome.[18]
Antigenic gift: Occurs when humans deliberately modify a virus's genome either in a lab setting or in order to make a bioweapon.
Influenza virus
The antigenic properties of influenza viruses are determined by both hemagglutinin and neuraminidase. Specific host proteases cleave the single peptide HA into two subunits HA1 and HA2. The virus becomes highly virulent if the amino acids at the cleavage sites are lipophilic. Selection pressure in the environment selects for antigenic changes in the antigen determinants of HA, that includes places undergoing adaptive evolution and in antigenic locations undergoing substitutions, which ultimately results in changes in the antigenicity of the virus. Glycosylation of HA does not correlate with either the antigenicity or the selection pressure.[19] Antigenic variation may be classified into two types, antigenic drift that results from a change in few amino acids and antigenic shift which is the outcome of acquiring new structural proteins. A new vaccine is required every year because influenza virus has the ability to undergo antigenic drift. Antigenic shift occurs periodically when the genes for structural proteins are acquired from other animal hosts resulting in a sudden dramatic change in viral genome. Recombination between segments that encode for hemagglutinin and neuraminidase of avian and human influenza virus segments have resulted in worldwide influenza epidemics called pandemics such as the Asian flu of 1957 when 3 genes from Eurasian avian viruses were acquired and underwent reassortment with 5 gene segments of the circulating human strains. Another example comes from the 1968 Hong Kong flu which acquired 2 genes by reassortment from Eurasian avian viruses with the 6 gene segments from circulating human strains.
Vaccination against influenza
After vaccination, IgG+ antibody-secreting plasma cells (ASCs) increase rapidly and reaches a maximum level at day 7 before returning to a minimum level at day 14. The influenza-specific memory B-cells reach their maxima at day 14–21. The secreted antibodies are specific to the vaccine virus. Further, most of the monoclonal antibodies isolated have binding affinities against HA and the remaining demonstrate affinity against NA, nucleoprotein (NP) and other antigens. These high affinity human monoclonal antibodies can be produced within a month after vaccination and because of their human origin, they will have very little, if any, antibody-related side-effects in humans. They can potentially be used to develop passive antibody therapy against influenza virus transmission.
Mapping antigenic evolution
The ability of an antiviral antibody to inhibit hemagglutination can be measured and used to generate a two-dimensional map using a process called antigenic cartography so that antigenic evolution can be visualized. These maps can show how changes in amino acids can alter the binding of an antibody to virus particle and help to analyze the pattern of genetic and antigenic evolution. Recent findings show that as a result of antibody-driven antigenic variation in one domain of the H1 hemagglutinin Sa site, a compensatory mutation in NA can result leading to NA antigenic variation. As a consequence, drug resistance develops to NA inhibitors. Such a phenomenon can mask the evolution of NA evolution in nature because the resistance to NA inhibitors could be due to antibody-driven, HA escape.[20]
HIV-1
The major challenge in controlling HIV-1 infection in the long term is immune escape. The extent and frequency to which an epitope will be targeted by a particular HLA allele differs from person-to-person. Moreover, as a consequence of immunodominance, an individual's CTL response is limited to a few epitopes of a specific HLA allele although six HLA class 1 alleles are expressed. Although the CTL response in the acute phase is directed against limited number of epitopes, the epitopic repertoire increases with time due to viral escape. Additionally amino acid co-evolution is a challenging issue that needs to be addressed. For example, a substitution in a particular site results in a secondary or compensatory mutation in another site. An invaluable discovery was that when a selective pressure is applied, the pattern of HIV-1 evolution can be predicted. In individuals who express a protective HLA B*27 allele, the first mutation that occurs in the Gag epitope KK10 is at position 6 from an L to an M and after several years there is a change in position 2 from a R to a K. Therefore, the knowledge of the predictability of the escape pathways can be utilized to design immunogens.[21] The region gp120 of HIV-1 Env which contacts CD4, its primary receptor, is functionally conserved and vulnerable to neutralizing antibodies such as monoclonal antibody b12. Recent findings show that resistance to neutralization by b12 was an outcome of substitutions that resided in the region proximal to CD4 contact surface. In this way the virus evades neutralization by b12 without affecting its binding to CD4.[22]
Flaviviruses
Flaviviridae is a family of viruses that encompasses well known viruses such as West Nile virus and Dengue virus. The genus Flavivirus has a prototypical envelope protein (E-protein) on its surface which serves as the target for virus neutralizing antibodies. E protein plays a role in binding to receptor and could play a role in evading the host immune system. It has three major antigenic domains namely A, B and C that correspond to the three structural domains II, III and I. Structural domain III is a putative receptor binding domain and antibodies against it neutralize the infectivity of flaviviruses. Mutations that lead to antigenic differences can be traced to the biochemical nature of the amino acid substitutions as well as the location of the mutation in the domain III. For example, substitutions at different amino acids results in varying levels of neutralization by antibodies. If mutation in a critical amino acid can dramatically alter neutralization by antibodies then WNV vaccines and diagnostic assays becomes difficult to rely on. Other flaviviruses that cause dengue, louping ill and yellow fever escape antibody neutralization via mutations in the domain III of the E protein.[23][24]
References
- Pays, Etienne; et al. (1983). "Gene conversion as a mechanism for antigenic variation in Trypanosomes". Cell. 34 (2): 371–381. doi:10.1016/0092-8674(83)90371-9. PMID 6616615. S2CID 688880.
- Lysnyansky, I.; Ron, Y.; Yogev, D. (2001). "Juxtaposition of an Active Promoter to vsp Genes via Site-Specific DNA Inversions Generates Antigenic Variation in Mycoplasma bovis". Journal of Bacteriology. 183 (19): 5698–5708. doi:10.1128/JB.183.19.5698-5708.2001. PMC 95462. PMID 11544233.
- Brunham, Robert C.; et al. (1993). "Bacterial Antigenic Variation, Host Immune Response, and Pathogen-Host Coevolution". Infection and Immunity. 61 (6): 2273–2276. doi:10.1128/IAI.61.6.2273-2276.1993. PMC 280844. PMID 8500868.
- Zhang, Jing-Ren; et al. (1997). "Antigenic Variation in Lyme Disease Borreliae by Promiscuous Recombination of VMP-like Sequence Cassettes". Cell. 89 (2): 275–285. doi:10.1016/S0092-8674(00)80206-8. PMID 9108482.
- Avery, S. V. (2006). "Microbial cell individuality and the underlying sources of heterogeneity". Nat Rev Microbiol. 4 (8): 577–87. doi:10.1038/nrmicro1460. PMID 16845428. S2CID 27337497.
- van der Woude, Marjan W.; et al. (2004). "Phase and Antigenic Variation in Bacteria". American Society for Microbiology. 17 (3): 581–611. doi:10.1128/CMR.17.3.581-611.2004. PMC 452554. PMID 15258095.
- Wisniewski-Dyé F; Vial L (2008). "Phase and antigenic variation mediated by genome modifications". Antonie van Leeuwenhoek. 94 (4): 493–515. doi:10.1007/s10482-008-9267-6. PMID 18663597. S2CID 25378695.
- Stockdale C; Swiderski MR; Barry JD; McCulloch R (2008). "Antigenic variation in Trypanosoma brucei: joining the DOTs". PLOS Biol. 6 (7): e185. doi:10.1371/journal.pbio.0060185. PMC 2486309. PMID 18666832.
- Hartley CL; McCulloch R (2008). "Trypanosoma brucei BRCA2 acts in antigenic variation and has undergone a recent expansion in BRC repeat number that is important during homologous recombination". Mol Microbiol. 68 (5): 1237–51. doi:10.1111/j.1365-2958.2008.06230.x. PMC 2408642. PMID 18430140.
- Lythgoe KA, Morrison LJ, Read AF, Barry JD (2007). "Parasite-intrinsic factors can explain ordered progression of trypanosome antigenic variation". Proceedings of the National Academy of Sciences. 104 (19): 8095–100. doi:10.1073/pnas.0606206104. PMC 1876577. PMID 17463092.
- Mideo, Nicole; Acosta-Serrano, Alvaro; Aebischer, Toni; Brown, Mark J.F.; Fenton, Andy; Friman, Ville-Petri; Restif, Olivier; Reece, Sarah E.; Webster, Joanne P.; Brown, Sam P. (2013). "Life in cells, hosts, and vectors: Parasite evolution across scales". Infection, Genetics and Evolution. Elsevier. 13: 344–347. doi:10.1016/j.meegid.2012.03.016. ISSN 1567-1348. PMID 22465537. S2CID 206881591.
- Kyes SA, Kraemer SM, Smith JD (2007). "Antigenic variation in Plasmodium falciparum: gene organization and regulation of the var multigene family". Eukaryot Cell. 6 (9): 1511–20. doi:10.1128/EC.00173-07. PMC 2043368. PMID 17644655.
- Scherf A, Hernandez-Rivas R, Buffet P, Bottius E, Benatar C, Pouvelle B, Gysin J, Lanzer M (1998). "Antigenic variation in malaria: in situ switching, relaxed and mutually exclusive transcription of var genes during intra-erythrocytic development in Plasmodium falciparum". EMBO J. 17 (18): 5418–26. doi:10.1093/emboj/17.18.5418. PMC 1170867. PMID 9736619.
- Kyes S, Christodoulou Z, Pinches R, Kriek N, Horrocks P, Newbold C (2007). "Plasmodium falciparum var gene expression is developmentally controlled at the level of RNA polymerase II-mediated transcription initiation". Mol Microbiol. 63 (4): 1237–47. doi:10.1111/j.1365-2958.2007.05587.x. PMID 17257309.
- Ralph SA, Scheidig-Benatar C, Scherf A (2005). "Antigenic variation in Plasmodium falciparum is associated with movement of var loci between subnuclear locations". Proc Natl Acad Sci U S A. 102 (15): 5414–9. doi:10.1073/pnas.0408883102. PMC 556247. PMID 15797990.
- Dufy, Siobain (March 4, 2008). "Rates of evolutionary change in viruses: patterns and determinants". Nature. 9 (4): 267–276. doi:10.1038/nrg2323. PMID 18319742. S2CID 10305325.
- Bean, William (3 December 1979). "Recombination of human influenza A viruses in nature". Nature. 284 (5757): 638–40. doi:10.1038/284638a0. PMID 7366737. S2CID 4368679.
- Lyttle, D J (Jan 1994). "Homologs of vascular endothelial growth factor are encoded by the poxvirus orf virus". Journal of Virology. 68 (1): 84–92. doi:10.1128/JVI.68.1.84-92.1994. PMC 236267. PMID 8254780.
- Chen, Jiezhong; Deng, Yi-Mo (2009). "Influenza virus antigenic variation, host antibody production and new approach to control epidemics". Virology Journal. 6: 30. doi:10.1186/1743-422X-6-30. PMC 2666653. PMID 19284639.
- Hensley, Scott E.; Das, Suman R.; Gibbs, James S.; Bailey, Adam L.; Schmidt, Loren M.; Bennink, Jack R.; Yewdell, Jonathan W. (2011). De La Torre, Juan C. (ed.). "Influenza A Virus Hemagglutinin Antibody Escape Promotes Neuraminidase Antigenic Variation and Drug Resistance". PLOS ONE. 6 (2): e15190. doi:10.1371/journal.pone.0015190. PMC 3043005. PMID 21364978.
- Carlson, JM; Brumme, ZL (2008). "HIV evolution in response to HLA-restricted CTL selection pressures: a population-based perspective". Microbes and Infection / Institut Pasteur. 10 (5): 455–61. doi:10.1016/j.micinf.2008.01.013. PMID 18407775.
- Li, H; Xu, CF; Blais, S; Wan, Q; Zhang, HT; Landry, SJ; Hioe, CE (2009). "Proximal glycans outside of the epitopes regulate the presentation of HIV-1 envelope gp120 helper epitopes". Journal of Immunology. 182 (10): 6369–78. doi:10.4049/jimmunol.0804287. PMC 2808118. PMID 19414790.
- Diamond, MS (2003). "Evasion of innate and adaptive immunity by flaviviruses". Immunology and Cell Biology. 81 (3): 196–206. doi:10.1046/j.1440-1711.2003.01157.x. PMID 12752684. S2CID 37056432.
- Li, Li; Barrett, Alan D.T.; Beasley, David W.C. (2005). "Differential expression of domain III neutralizing epitopes on the envelope proteins of West Nile virus strains". Virology. 335 (1): 99–105. doi:10.1016/j.virol.2005.02.011. PMID 15823609.