Somatic evolution in cancer
Somatic evolution is the accumulation of mutations and epimutations in somatic cells (the cells of a body, as opposed to germ plasm and stem cells) during a lifetime, and the effects of those mutations and epimutations on the fitness of those cells. This evolutionary process has first been shown by the studies of Bert Vogelstein in colon cancer. Somatic evolution is important in the process of aging as well as the development of some diseases, including cancer.
Natural selection in cancer
Cells in pre-malignant and malignant neoplasms (tumors) evolve by natural selection.[1][2] This accounts for how cancer develops from normal tissue and why it has been difficult to cure. There are three necessary and sufficient conditions for natural selection, all of which are met in a neoplasm:
- There must be variation in the population. Neoplasms are mosaics of different mutant cells with both genetic and epigenetic changes that distinguish them from normal cells.
- The variable traits must be heritable. When a cancer cell divides, both daughter cells inherit the genetic and epigenetic abnormalities of the parent cell, and may also acquire new genetic and epigenetic abnormalities in the process of cellular reproduction.
- That variation must affect survival or reproduction (fitness). While many of the genetic and epigenetic abnormalities in neoplasms are probably neutral evolution, many have been shown to increase the proliferation of the mutant cells, or decrease their rate of death (apoptosis).[3] (See Hallmarks below)
Cells in neoplasms compete for resources, such as oxygen and glucose, as well as space. Thus, a cell that acquires a mutation that increases its fitness will generate more daughter cells than competitor cells that lack that mutation. In this way, a population of mutant cells, called a clone, can expand in the neoplasm. Clonal expansion is the signature of natural selection in cancer.
Cancer therapies act as a form of artificial selection, killing sensitive cancer cells, but leaving behind resistant cells. Often the tumor will regrow from those resistant cells, the patient will relapse, and the therapy that had been previously used will no longer kill the cancer cells. This selection for resistance is similar to the repeatedly spraying crops with a pesticide and selecting for resistant pests until the pesticide is no longer effective.
Evolution in complex biological systems
Modern descriptions of biological evolution will typically elaborate on major contributing factors to evolution such as the formation of local micro-environments, mutational robustness, molecular degeneracy, and cryptic genetic variation.[4] Many of these contributing factors in evolution have been isolated and described for cancer.[5]
Multilevel selection
Cancer is a classic example of what evolutionary biologists call multilevel selection: at the level of the organism, cancer is usually fatal so there is selection for genes and the organization of tissues[6][7] that suppress cancer. At the level of the cell, there is selection for increased cell proliferation and survival, such that a mutant cell that acquires one of the hallmarks of cancer[3] (see below), will have a competitive advantage over cells that have not acquired the hallmark. Thus, at the level of the cell there is selection for cancer.
History
Pre-Nowell & Cairns
The earliest ideas about neoplastic evolution come from Boveri[8] who proposed that tumors originated in chromosomal abnormalities passed on to daughter cells. In the decades that followed, cancer was recognized as having a clonal origin associated with chromosomal aberrations.[9][10][11][12]
Early mathematical modeling of cancer, by Armitage and Doll, set the stage for the future development of the somatic evolutionary theory of cancer. Armitage and Doll explained the cancer incidence data, as a function of age, as a process of the sequential accumulation of somatic mutations (or other rate limiting steps).[13]
Advances in cytogenetics facilitated discovery of chromosome abnormalities in neoplasms, including the Philadelphia chromosome in chronic myelogenous leukemia[14] and translocations in acute myeloblastic leukemia.[15] Sequences of karyotypes replacing one another in a tumor were observed as it progressed.[16][17][18] Researchers hypothesized that cancer evolves in a sequence of chromosomal mutations and selection[6][17][19][20] and that therapy may further select clones.[12]
Knudson, Cairns, and Nowell
In 1971, Knudson published the 2-hit hypothesis for mutation and cancer based on statistical analysis of inherited and sporadic cases of retinoblastoma.[21] He postulated that retinoblastoma developed as a consequence of two mutations; one of which could be inherited or somatic followed by a second somatic mutation. Cytogenetic studies localized the region to the long arm of chromosome 13, and molecular genetic studies demonstrated that tumorigenesis was associated with chromosomal mechanisms, such as mitotic recombination or non-disjunction, that could lead to homozygosity of the mutation.[22] The retinoblastoma gene was the first tumor suppressor gene to be cloned in 1986.
Cairns hypothesized a different, but complementary, mechanism of tumor suppression in 1975 based on tissue architecture to protect against selection of variant somatic cells with increased fitness in proliferating epithelial populations, such as the intestine and other epithelial organs.[6] He postulated that this could be accomplished by restricting the number of stem cells for example at the base of intestinal crypts and restraining the opportunities for competition between cells by shedding differentiated intestinal cells into the gut. The essential predictions of this model have been confirmed although mutations in some tumor suppressor genes, including CDKN2A (p16), predispose to clonal expansions that encompass large numbers of crypts in some conditions such as Barrett's esophagus. He also postulated an immortal DNA strand that is discussed at Immortal DNA strand hypothesis.
Nowell synthesized the evolutionary view of cancer in 1976 as a process of genetic instability and natural selection.[1] Most of the alterations that occur are deleterious for the cell, and those clones will tend to go extinct, but occasional selectively advantageous mutations arise that lead to clonal expansions. This theory predicts a unique genetic composition in each neoplasm due to the random process of mutations, genetic polymorphisms in the human population, and differences in the selection pressures of the neoplasm's microenvironment. Interventions are predicted to have varying results in different patients. What is more important, the theory predicts the emergence of resistant clones under the selective pressures of therapy. Since 1976, researchers have identified clonal expansions[23][24][25][26][27][28] and genetic heterogeneity[29][30][31][32][33][34] within many different types of neoplasms.
Somatic evolution in progression
Genetic heterogeneity in neoplasms
There are multiple levels of genetic heterogeneity associated with cancer, including single nucleotide polymorphism (SNP),[35] sequence mutations,[30] Microsatellite shifts[29] and instability,[36] loss of heterozygosity (LOH),[34] Copy number variation (detected both by comparative genomic hybridization (CGH),[31] and array CGH,[37]) and karyotypic variations including chromosome structural aberrations and aneuploidy.[32][33][38][39][40] Studies of this issue have focused mainly at the gene mutation level, as copy number variation, LOH and specific chromosomal translocations are explained in the context of gene mutation. It is thus necessary to integrate multiple levels of genetic variation in the context of complex system and multilevel selection.
System instability is a major contributing factor for genetic heterogeneity.[41] For the majority of cancers, genome instability is reflected in a large frequency of mutations in the whole genome DNA sequence (not just the protein coding regions that are only 1.5% of the genome[42]). In whole genome sequencing of different types of cancers, large numbers of mutations were found in two breast cancers (about 20,000 point mutations[43]), 25 melanomas (9,000 to 333,000 point mutations[44]) and a lung cancer (50,000 point mutations and 54,000 small additions and deletions[45]). Genome instability is also referred to as an enabling characteristic for achieving endpoints of cancer evolution.[3]
Many of the somatic evolutionary studies have traditionally been focused on clonal expansion, as recurrent types of changes can be traced to illustrate the evolutionary path based on available methods. Recent studies from both direct DNA sequencing and karyotype analysis illustrate the importance of the high level of heterogeneity in somatic evolution. For the formation of solid tumors, there is an involvement of multiple cycles of clonal and non-clonal expansion.[39][46] Even at the typical clonal expansion phase, there are significant levels of heterogeneity within the cell population, however, most are under-detected when mixed populations of cells are used for molecular analysis. In solid tumors, a majority of gene mutations are not recurrent types,[47] and neither are the karyotypes.[39][41] These analyses offer an explanation for the findings that there are no common mutations shared by most cancers.[48]
Somatic evolution by epigenetics
The state of a cell may be changed epigenetically, in addition to genetic alterations. The best-understood epigenetic alterations in tumors are the silencing or expression of genes by changes in the methylation of CG pairs of nucleotides in the promoter regions of the genes. These methylation patterns are copied to the new chromosomes when cells replicate their genomes and so methylation alterations are heritable and subject to natural selection. Methylation changes are thought to occur more frequently than mutations in the DNA, and so may account for many of the changes during neoplastic progression (the process by which normal tissue becomes cancerous), in particular in the early stages. For instance, when loss of expression of the DNA repair protein MGMT occurs in a colon cancer, it is caused by a mutation only about 4% of the time, while in most cases the loss is due to methylation of its promoter region.[49] Similarly, when loss of expression of the DNA repair protein PMS2 occurs in colon cancer, it is caused by a mutation about 5% of the time, while in most cases loss of expression is due to methylation of the promoter of its pairing partner MLH1 (PMS2 is unstable in the absence of MLH1).[50] Epigenetic changes in progression interact with genetic changes. For example, epigenetic silencing of genes responsible for the repair of mispairs or damages in the DNA (e.g. MLH1 or MSH2) results in an increase of genetic mutations.
Deficiency of DNA repair proteins PMS2, MLH1, MSH2, MSH3, MSH6 or BRCA2 can cause up to 100-fold increases in mutation frequency[51][52][53] Epigenetic deficiencies in DNA repair gene protein expression have been found in many cancers, though not all deficiencies have been evaluated in all cancers. Epigeneticically deficient DNA repair proteins include BRCA1, WRN, MGMT, MLH1, MSH2, ERCC1, PMS2, XPF, P53, PCNA and OGG1, and these are found to be deficient at frequencies of 13% to 100% in different cancers. (Also see Frequencies of epimutations in DNA repair genes.)
In addition to well studied epigenetic promoter methylation, more recently there have been substantial findings of epigenetic alterations in cancer due to changes in histone and chromatin architecture and alterations in the expression of microRNAs (microRNAs either cause degradation of messenger RNAs or block their translation)[54] For instance, hypomethylation of the promoter for microRNA miR-155 increases expression of miR-155, and this increased miR-155 targets DNA repair genes MLH1, MSH2 and MSH6, causing each of them to have reduced expression.[55]
In cancers, loss of expression of genes occurs about 10 times more frequently by transcription silencing (caused by somatically heritable promoter hypermethylation of CpG islands) than by mutations. As Vogelstein et al. point out, in a colorectal cancer there are usually about 3 to 6 driver mutations and 33 to 66 hitchhiker or passenger mutations.[56] In contrast, in colon tumors compared to adjacent normal-appearing colonic mucosa, there are about 600 to 800 somatically heritable heavily methylated CpG islands in promoters of genes in the tumors while these CpG islands are not methylated in the adjacent mucosa.[57][58][59]
Methylation of the cytosine of CpG dinucleotides is a somatically heritable and conserved regulatory mark that is generally associated with transcriptional repression. CpG islands keep their overall un-methylated state (or methylated state) extremely stably through multiple cell generations.[60]
Clonal expansions
One common feature of neoplastic progression is the expansion of a clone with a genetic or epigenetic alteration. This may be a matter of chance, but is more likely due to the expanding clone having a competitive advantage (either a reproductive or survival advantage) over other cells in the tissue. Since clones often have many genetic and epigenetic alterations in their genomes, it is often not clear which of those alterations cause a reproductive or survival advantage and which other alterations are simply hitchhikers or passenger mutations (see Glossary below) on the clonal expansion.
Clonal expansions are most often associated with the loss of the p53 (TP53) or p16 (CDKN2A/INK4a) tumor suppressor genes. In lung cancer, a clone with a p53 mutation was observed to have spread over the surface of one entire lung and into the other lung.[27] In bladder cancer, clones with loss of p16 were observed to have spread over the entire surface of the bladder.[61][62] Likewise, large expansions of clones with loss of p16 have been observed in the oral cavity[24] and in Barrett's esophagus.[25] Clonal expansions associated with inactivation of p53 have also appear in skin,[23][63] Barrett's esophagus,[25] brain,[64] and kidney.[65] Further clonal expansions have been observed in the stomach,[66] bladder,[67] colon,[68] lung,[69] hematopoietic (blood) cells,[70] and prostate.[71]
These clonal expansions are important for at least two reasons. First, they generate a large target population of mutant cells and so increase the probability that the multiple mutations necessary to cause cancer will be acquired within that clone. Second, in at least one case, the size of the clone with loss of p53 has been associated with an increased risk of a pre-malignant tumor becoming cancerous.[72] It is thought that the process of developing cancer involves successive waves of clonal expansions within the tumor.[73]
Field defects
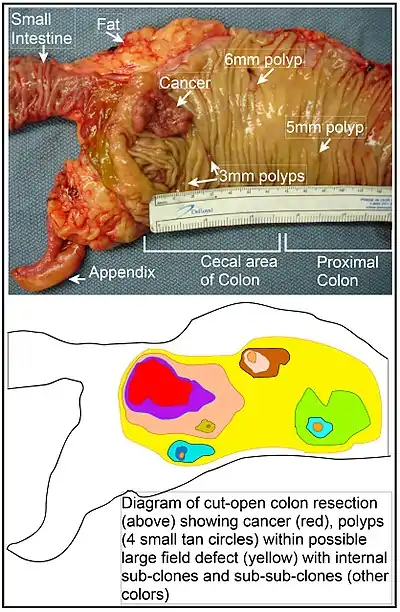
The term "field cancerization" was first used in 1953 to describe an area or "field" of epithelium that has been preconditioned by (at that time) largely unknown processes so as to predispose it towards development of cancer.[74] Since then, the terms "field cancerization" and "field defect" have been used to describe pre-malignant tissue in which new cancers are likely to arise. Field defects, for example, have been identified in most of the major areas subject to tumorigenesis in the gastrointestinal (GI) tract.[75] Cancers of the GI tract that are shown to be due, to some extent, to field defects include head and neck squamous cell carcinoma (HNSCC), oropharyngeal/laryngeal cancer, esophageal adenocarcinoma and esophageal squamous-cell carcinoma, gastric cancer, bile duct cancer, pancreatic cancer, small intestine cancer and colon cancer.
In the colon, a field defect probably arises by natural selection of a mutant or epigenetically altered cell among the stem cells at the base of one of the intestinal crypts on the inside surface of the colon. A mutant or epigenetically altered stem cell, if it has a selective advantage, could replace the other nearby stem cells by natural selection. This can cause a patch of abnormal tissue, or field defect. The figure in this section includes a photo of a freshly resected and lengthwise-opened segment of the colon that may represent a large field defect in which there is a colon cancer and four polyps. The four polyps, in addition to the cancer, may represent sub-clones with proliferative advantages.
The sequence of events giving rise to this possible field defect are indicated below the photo. The schematic diagram shows a large area in yellow indicating a large patch of mutant or epigenetically altered cells that formed by clonal expansion of an initial cell based on a selective advantage. Within this first large patch, a second such mutation or epigenetic alteration may have occurred so that a given stem cell acquired an additional selective advantage compared to the other stem cells within the patch, and this altered stem cell expanded clonally forming a secondary patch, or sub-clone, within the original patch. This is indicated in the diagram by four smaller patches of different colors within the large yellow original area. Within these new patches (sub-clones), the process may have been repeated multiple times, indicated by the still smaller patches within the four secondary patches (with still different colors in the diagram) which clonally expanded, until a stem cell arose that generated either small polyps (which may be benign neoplasms) or else a malignant neoplasm (cancer). These neoplasms are also indicated, in the diagram below the photo, by 4 small tan circles (polyps) and a larger red area (cancer). The cancer in the photo occurred in the cecal area of the colon, where the colon joins the small intestine (labeled) and where the appendix occurs (labeled). The fat in the photo is external to the outer wall of the colon. In the segment of colon shown here, the colon was cut open lengthwise to expose the inner surface of the colon and to display the cancer and polyps occurring within the inner epithelial lining of the colon.
Phylogenetic analyses
Phylogenetics may be applied to cells in tumors to reveal the evolutionary relationships between cells, just as it is used to reveal evolutionary relationships between organisms and species. Shibata, Tavare and colleagues have exploited this to estimate the time between the initiation of a tumor and its detection in the clinic.[29] Louhelainen et al. have used parsimony to reconstruct the relationships between biopsy samples based on loss of heterozygosity.[76] Phylogenetic trees should not be confused with oncogenetic trees,[77] which represent the common sequences of genetic events during neoplastic progression and do not represent the relationships of common ancestry that are essential to a phylogeny. For an up-to-date review in this field, see Bast 2012.[78]
Adaptive landscapes
An adaptive landscape is a hypothetical topological landscape upon which evolution is envisioned to take place. It is similar to Wright's fitness landscape[79][80] in which the location of each point represents the genotype of an organism and the altitude represents the fitness of that organism in the current environment. However, unlike Wright's rigid landscape, the adaptive landscape is pliable. It readily changes shape with changes in population densities and survival/reproductive strategies used within and among the various species.
Wright's shifting balance theory of evolution combines genetic drift (random sampling error in the transmission of genes) and natural selection to explain how multiple peaks on a fitness landscape could be occupied or how a population can achieve a higher peak on this landscape. This theory, based on the assumption of density-dependent selection as the principal forms of selection, results in a fitness landscape that is relatively rigid. A rigid landscape is one that does not change in response to even large changes in the position and composition of strategies along the landscape.
In contrast to the fitness landscape, the adaptive landscape is constructed assuming that both density and frequency-dependent selection is involved (selection is frequency-dependant when the fitness of a species depends not only on that species strategy but also on the strategy of all other species). As such, the shape of the adaptive landscape can change drastically in response to even small changes in strategies and densities.[81]
The flexibility of adaptive landscapes provide several ways for natural selection to cross valleys and occupy multiple peaks without having to make large changes in their strategies. Within the context of differential or difference equation models for population dynamics, an adaptive landscape may actually be constructed using a fitness generating function.[82] If a given species is able to evolve, it will, over time, "climb" the adaptive landscape toward a fitness peak through gradual changes in its mean phenotype according to a strategy dynamic that involves the slope of the adaptive landscape. Because the adaptive landscape is not rigid and can change shape during the evolutionary process, it is possible that a species may be driven to maximum, minimum, or saddle point on the adaptive landscape. A population at a global maximum on the adaptive landscape corresponds an evolutionarily stable strategy (ESS) and will become dominant, driving all others toward extinction. Populations at a minimum or saddle point are not resistant to invasion, so that the introduction of a slightly different mutant strain may continue the evolutionary process toward unoccupied local maxima.
The adaptive landscape provides a useful tool for studying somatic evolution as it can describe the process of how a mutant cell evolves from a small tumor to an invasive cancer. Understanding this process in terms of the adaptive landscape may lead to the control of cancer through external manipulation of the shape of the landscape.[83][84]
The Hallmarks of Cancer as evolutionary adaptations in a neoplasm
In their landmark paper, The Hallmarks of Cancer,[3] Hanahan and Weinberg suggest that cancer can be described by a small number of underlying principles, despite the complexities of the disease. The authors describe how tumor progression proceeds via a process analogous to Darwinian evolution, where each genetic change confers a growth advantage to the cell. These genetic changes can be grouped into six "hallmarks", which drive a population of normal cells to become a cancer. The six hallmarks are:
- self-sufficiency in growth signals
- insensitivity to antigrowth signals
- evasion of apoptosis
- limitless replicative potential
- sustained angiogenesis, and
- tissue invasion and metastasis.
Genetic instability is defined as an "enabling characteristic" that facilitates the acquisition of other mutations due to defects in DNA repair.
The hallmark "self-sufficiency in growth signals" describes the observation that tumor cells produce many of their own growth signals and thereby no longer rely on proliferation signals from the micro-environment. Normal cells are maintained in a nondividing state by antigrowth signals, which cancer cells learn to evade through genetic changes producing "insensitivity to antigrowth signals". A normal cell initiates programmed cell death (apoptosis) in response to signals such as DNA damage, oncogene overexpression, and survival factor insufficiency, but a cancer cell learns to "evade apoptosis", leading to the accumulation of aberrant cells. Most mammalian cells can replicate a limited number of times due to progressive shortening of telomeres; virtually all malignant cancer cells gain an ability to maintain their telomeres, conferring "limitless replicative potential". As cells cannot survive at distances of more than 100 μm from a blood supply, cancer cells must initiate the formation of new blood vessels to support their growth via the process of "sustained angiogenesis". During the development of most cancers, primary tumor cells acquire the ability to undergo "invasion and metastasis" whereby they migrate into the surrounding tissue and travel to distant sites in the body, forming secondary tumors.
The pathways that cells take toward becoming malignant cancers are variable, and the order in which the hallmarks are acquired can vary from tumor to tumor. The early genetic events in tumorigenesis are difficult to measure clinically, but can be simulated according to known biology.[85] Macroscopic tumors are now beginning to be described in terms of their underlying genetic changes, providing additional data to refine the framework described in The Hallmarks of Cancer.
Clonal evolution and cancer stem cells
Monoclonal theory of cancer origin
The theory about the monoclonal origin of cancer states that, in general, neoplasms arise from a single cell of origin.[1] While it is possible that certain carcinogens may mutate more than one cell at once, the tumor mass usually represents progeny of a single cell, or very few cells.[1] A series of mutations is required in the process of carcinogenesis for a cell to transition from being normal to pre-malignant and then to a cancer cell.[86] The mutated genes usually belong to classes of caretaker, gatekeeper, landscaper or several other genes. Mutation ultimately leads to acquisition of the ten hallmarks of cancer.
Cancer stem cells
The first malignant cell, that gives rise to the tumor, is often labeled a cancer stem cell.[87]
The cancer stem-cell hypothesis relies on the fact that a lot of tumors are heterogeneous – the cells in the tumor vary by phenotype and functions.[87][88][89] Current research shows that in many cancers there is apparent hierarchy among cells.[87][88][89] in general, there is a small population of cells in the tumor – about 0.2%–1%[88] – that exhibits stem cell-like properties. These cells have the ability to give rise to a variety of cells in tumor tissue, self-renew indefinitely, and upon transfer can form new tumors. According to the hypothesis, cancer stem cells are the only cells capable of tumorigenesis – initiation of a new tumor.[87] Cancer stem cell hypothesis might explain such phenomena as metastasis and remission.
The monoclonal model of cancer and the cancer stem-cell model are not mutually exclusive.[87] Cancer stem cell arises by clonal evolution as a result of selection for the cell with the highest fitness in the neoplasm. This way, the heterogeneous nature of neoplasm can be explained by two processes – clonal evolution, or the hierarchical differentiation of cells, regulated by cancer stem cells.[87] All cancers arise as a result of somatic evolution, but only some of them fit the cancer stem cell hypothesis.[87] The evolutionary processes do not cease when a population of cancer stem cells arises in a tumor. Cancer treatment drugs pose a strong selective force on all types of cells in tumors, including cancer stem cells, which would be forced to evolve resistance to the treatment. Cancer stem cells do not always have to have the highest resistance among the cells in the tumor to survive chemotherapy and re-emerge afterwards. The surviving cells might be in a special microenvironment, which protects them from adverse effects of treatment.[87]
It is currently unclear as to whether cancer stem cells arise from adult stem cell transformation, a maturation arrest of progenitor cells, or as a result of dedifferentiation of mature cells.[88]
Somatic evolution in therapeutic resistance
Therapeutic resistance has been observed in virtually every form of therapy, from the beginning of cancer therapy.[90] In most cases, therapies appear to select for mutations in the genes or pathways targeted by the drug.
Resistance to methotrexate
Some of the first evidence for a genetic basis of acquired therapeutic resistance came from studies of methotrexate. Methotrexate inhibits the dihydrofolate reductase (DHFR) gene. However, methotrexate therapy appears to select for cells with extra copies (amplification) of DHFR, which are resistant to methotrexate. This was seen in both cell culture[91] and samples from tumors in patients that had been treated with methotrexate.[92][93][94][95]
Resistance to 5-fluorouracil
A common cytotoxic chemotherapy used in a variety of cancers, 5-fluorouracil (5-FU), targets the TYMS pathway and resistance can evolve through the evolution of extra copies of TYMS, thereby diluting the drug's effect.[96]
Resistance to BCR-ABL targeting drugs
In the case of Gleevec (Imatinib), which targets the BCR-ABL fusion gene in chronic myeloid leukemia, resistance often develops through a mutation that changes the shape of the binding site of the drug.[97][98] Sequential application of drugs can lead to the sequential evolution of resistance mutations to each drug in turn.[99]
Gleevec is not as selective as was originally thought. It turns out that it targets other tyrosine kinase genes and can be used to control gastrointestinal stromal tumors (GISTs) that are driven by mutations in c-KIT. However, patients with GIST sometimes relapse with additional mutations in c-KIT that make the cancer cells resistant to Gleevec.[100][101]
Resistance to EGFR targeting drugs
Gefitinib(Iressa) and Erlotinib (Tarceva) are epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors used for non-small cell lung cancer patients whose tumors have somatic mutations in EGFR. However, most patients' tumors eventually become resistant to these drugs. Two major mechanisms of acquired resistance have been discovered in patients who have developed clinical resistance to Gefitinib or Erlotinib:[102] point mutations in the EGFR gene targeted by the drugs,[103] and amplification of MET, another receptor tyrosine kinase, which can bypass EGFR to activate downstream signaling in the cell. In an initial study, 22% of tumors with acquired resistance to Gefitinib or Erlotinib had MET amplification.[104] To address these issues, clinical trials are currently assessing irreversible EGFR inhibitors (which inhibit growth even in cell lines with mutations in EGFR), the combination of EGFR and MET kinase inhibitors, and Hsp90 inhibitors (EGFR and MET both require Hsp90 proteins to fold properly). In addition, taking repeated tumor biopsies from patients as they develop resistance to these drugs would help to understand the tumor dynamics.
Resistance to selective estrogen receptor modulator drugs
Selective estrogen receptor modulators (SERMs) are a commonly used adjuvant therapy in estrogen-receptor positive (ERα+) breast cancer and a preventive treatment for women at high risk of the disease. There are several possible mechanisms of SERM resistance, though the relative clinical importance of each is debated. These include:[105][106]
- Loss of estrogen receptor alpha (ERα)[107]
- Although this may be a mechanism of resistance in a minority of women, most ERα+ tumors that become resistant to SERMS remain ERα+[108]
- Increased relative expression of ERβ compared to ERα
- Interference/cross-talk with growth factor signaling pathways such as EGFR/HER2
- Mutations in estrogen receptors
- Alterations in co-regulatory proteins
- Interactions between the SERM, ER, and co-regulatory proteins may influence whether the SERM acts as an estrogen antagonist or as an estrogen agonist.
- Reduced metabolic activation of tamoxifen[109]
- Polymorphisms in CYP2D6 show variable rates of conversion of tamoxifen to its activated, anti-estrogenic form.[110]
Resistance to anti-androgen therapy
Most prostate cancers derive from cells that are stimulated to proliferate by androgens. Most prostate cancer therapies are therefore based on removing or blocking androgens. Mutations in the androgen receptor (AR) have been observed in anti-androgen resistant prostate cancer that makes the AR hypersensitive to the low levels of androgens that remain after therapy.[111] Likewise, extra copies of the AR gene (amplification) have been observed in anti-androgen resistant prostate cancer.[112] These additional copies of the gene are thought to make the cell hypersensitive to low levels of androgens and so allow them to proliferate under anti-androgen therapy.
Resistance to radiotherapy
Resistance to radiotherapy is also commonly observed. However, to date, comparisons of malignant tissue before and after radiotherapy have not been done to identify genetic and epigenetic changes selected by exposure to radiation. In gliomas, a form of brain cancer, radiation therapy appears to select for stem cells,[113][114] though it is unclear if the tumor returns to the pre-therapy proportion of cancer stem cells after therapy or if radiotherapy selects for an alteration that keeps the glioma cells in the stem cell state.
Harnessing evolution in therapeutics
Cancer drugs and therapies commonly used today are evolutionary inert and represent a strong selection force, which leads to drug resistance.[115] A possible way to avoid that is to use a treatment agent that would co-evolve alongside cancer cells.
Anoxic bacteria
Anoxic bacteria could be used as competitors or predators in hypoxic environments within tumors.[115] Scientists have been interested in the idea of using anoxic bacteria for over 150 years, but until recently there has been little progress in that field. According to Jain and Forbes, several requirements have to be met by the cells to qualify as efficient anticancer bacterium:[116]
- The bacterium cannot be toxic to the host.
- Its population should be restricted to the tumor mass.
- It should be able to disperse evenly throughout the neoplasm.
- At the end of the treatment bacterium should be easily eliminated from the host.
- It should not be causing severe immune response.
- It should be able to cause tumor cells death through competition for nutrients.
In the process of the treatment, cancer cells are most likely to evolve some form of resistance to the bacterial treatment. However, being a living organism, bacteria would coevolve with tumor cells, potentially eliminating the possibility of resistance.[116]
Possible limitations
Since bacteria prefer an anoxic environment, they are not efficient at eliminating cells on the periphery of the tumor, where oxygen supply is efficient. A combination of bacterial treatment with chemical drugs will increase chances of destroying the tumor.[116]
Oncolytic viruses
Oncolytic viruses are engineered to infect cancerous cells. Limitations of that method include immune response to the virus and the possibility of the virus evolving into a pathogen.[115]
Natural selection
By manipulating the tumor environment, it is possible to create favorable conditions for the cells with least resistance to chemotherapy drugs to become more fit and outcompete the rest of the population. The chemotherapy, administered directly after, should wipe out the predominant tumor cells.[115]
Glossary
Mapping between common terms from cancer biology and evolutionary biology:
- Driver mutation — a mutation that gives a selective advantage to a clone in its microenvironment, through either increasing its survival or reproduction. Driver mutations tend to cause clonal expansions.
- Passenger mutation — a mutation that has no effect on the fitness of a clone but may be associated with a clonal expansion because it occurs in the same genome with a driver mutation. This is known as a hitchhiker in evolutionary biology.
- Clone — a set of cells that all descend from a common ancestor cell. A clone is usually distinguished through inheritance of a distinctive genetic lesion (mutation) that occurred in the ancestor cell.
- Neoplastic progression — the somatic evolutionary process by which normal tissue changes into malignant (cancerous) tissue.
See also
- Tumour heterogeneity
- Cancer
- Cancer research
- Carcinogenesis
- Natural selection
References
- Nowell PC (October 1976). "The clonal evolution of tumor cell populations". Science. 194 (4260): 23–28. Bibcode:1976Sci...194...23N. doi:10.1126/science.959840. PMID 959840.
- Merlo LM, Pepper JW, Reid BJ, Maley CC (December 2006). "Cancer as an evolutionary and ecological process". Nature Reviews. Cancer. 6 (12): 924–935. doi:10.1038/nrc2013. PMID 17109012. S2CID 8040576.
- Hanahan D, Weinberg RA (January 2000). "The hallmarks of cancer". Cell. 100 (1): 57–70. doi:10.1016/S0092-8674(00)81683-9. PMID 10647931. S2CID 1478778.
- Whitacre JM (2011). "Genetic and environment-induced pathways to innovation: on the possibility of a universal relationship between robustness and adaptation in complex biological systems". Evolutionary Ecology. 25 (5): 965–975. doi:10.1007/s10682-011-9464-z.
- Tian T, Olson S, Whitacre JM, Harding A (January 2011). "The origins of cancer robustness and evolvability" (PDF). Integrative Biology. 3 (1): 17–30. doi:10.1039/c0ib00046a. PMID 20944865.
- Cairns J (May 1975). "Mutation selection and the natural history of cancer". Nature. 255 (5505): 197–200. Bibcode:1975Natur.255..197C. doi:10.1038/255197a0. PMID 1143315. S2CID 4216433.
- Pepper JW, Sprouffske K, Maley CC (December 2007). "Animal cell differentiation patterns suppress somatic evolution". PLOS Computational Biology. 3 (12): e250. Bibcode:2007PLSCB...3..250P. doi:10.1371/journal.pcbi.0030250. PMC 2134960. PMID 18085819. Also see commentary
- Manchester KL (October 1995). "Theodor Boveri and the origin of malignant tumours". Trends in Cell Biology. 5 (10): 384–387. doi:10.1016/S0962-8924(00)89080-7. PMID 14732055.
- Makino S (March 1956). "Further evidence favoring the concept of the stem cell in ascites tumors of rats". Annals of the New York Academy of Sciences. 63 (5): 818–830. Bibcode:1956NYASA..63..818M. doi:10.1111/j.1749-6632.1956.tb50894.x. PMID 13314436. S2CID 28319058.
- Hauschka TS (September 1961). "The chromosomes in ontogeny and oncogeny". Cancer Research. 21: 957–974. PMID 13712320.
- Levan A, Biesele JJ (September 1958). "Role of chromosomes in cancerogenesis, as studied in serial tissue culture of mammalian cells". Annals of the New York Academy of Sciences. 71 (6): 1022–1053. Bibcode:1958NYASA..71.1022L. doi:10.1111/j.1749-6632.1958.tb46820.x. PMID 13583868. Archived from the original on 2013-01-05.
- de Grouchy J, de Nava C (August 1968). "A chromosomal theory of carcinogenesis". Annals of Internal Medicine. 69 (2): 381–391. doi:10.7326/0003-4819-69-2-381. PMID 5243847.
- Armitage P, Doll R (March 1954). "The age distribution of cancer and a multi-stage theory of carcinogenesis". British Journal of Cancer. 8 (1): 1–12. doi:10.1038/bjc.1954.1. PMC 2007940. PMID 13172380.
- Nowell PC, Hungerford DA (July 1960). "Chromosome studies on normal and leukemic human leukocytes". Journal of the National Cancer Institute. 25: 85–109. doi:10.1093/jnci/25.1.85. PMID 14427847.
- Rowley JD (June 1973). "Identificaton of a translocation with quinacrine fluorescence in a patient with acute leukemia". Annales de Génétique. 16 (2): 109–112. PMID 4125056.
- Ford CE, Clarke CM (1963). "Cytogenetic evidence of clonal proliferation in primary reticular neoplasms". Proceedings. Canadian Cancer Conference. 5: 129–146. PMID 14278854.
- Yosida TH (1966). "Relation between Chromosomal Alteration and Development of Tumors". Japanese Journal of Genetics. 41 (6): 439–51. doi:10.1266/jjg.41.439.
- de Grouchy J, de Nava C, Cantu JM, Bilski-Pasquier G, Bousser J (September 1966). "Models for clonal evolutions: a study of chronic myelogenous leukemia". American Journal of Human Genetics. 18 (5): 485–503. PMC 1706184. PMID 5224748.
- de Grouchy J (January 1973). "Cancer and the evolution of species: a ransom". Biomédicine. 18 (1): 6–8. PMID 4197290.
- Ryser HJ (September 1971). "Chemical carcinogenesis". The New England Journal of Medicine. 285 (13): 721–734. doi:10.1056/NEJM197109232851305. PMID 4942982.
- Knudson AG (April 1971). "Mutation and cancer: statistical study of retinoblastoma". Proceedings of the National Academy of Sciences of the United States of America. 68 (4): 820–823. Bibcode:1971PNAS...68..820K. doi:10.1073/pnas.68.4.820. PMC 389051. PMID 5279523.
- Cavenee WK, Dryja TP, Phillips RA, Benedict WF, Godbout R, Gallie BL, et al. (1983). "Expression of recessive alleles by chromosomal mechanisms in retinoblastoma". Nature. 305 (5937): 779–784. Bibcode:1983Natur.305..779C. doi:10.1038/305779a0. PMID 6633649. S2CID 4248936.
- Brash DE, Zhang W, Grossman D, Takeuchi S (April 2005). "Colonization of adjacent stem cell compartments by mutant keratinocytes". Seminars in Cancer Biology. 15 (2): 97–102. doi:10.1016/j.semcancer.2004.08.006. PMID 15652454.
- Braakhuis BJ, Leemans CR, Brakenhoff RH (April 2005). "Expanding fields of genetically altered cells in head and neck squamous carcinogenesis". Seminars in Cancer Biology. 15 (2): 113–120. doi:10.1016/j.semcancer.2004.08.004. PMID 15652456.
- Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Reid BJ (May 2004). "Selectively advantageous mutations and hitchhikers in neoplasms: p16 lesions are selected in Barrett's esophagus". Cancer Research. 64 (10): 3414–3427. doi:10.1158/0008-5472.CAN-03-3249. PMID 15150093.
- Habuchi T (August 2005). "Origin of multifocal carcinomas of the bladder and upper urinary tract: molecular analysis and clinical implications". International Journal of Urology. 12 (8): 709–716. doi:10.1111/j.1442-2042.2005.01155.x. PMID 16174043. S2CID 30176505.
- Franklin WA, Gazdar AF, Haney J, Wistuba II, La Rosa FG, Kennedy T, et al. (October 1997). "Widely dispersed p53 mutation in respiratory epithelium. A novel mechanism for field carcinogenesis". The Journal of Clinical Investigation. 100 (8): 2133–2137. doi:10.1172/JCI119748. PMC 508406. PMID 9329980.
- Brentnall TA, Crispin DA, Rabinovitch PS, Haggitt RC, Rubin CE, Stevens AC, Burmer GC (August 1994). "Mutations in the p53 gene: an early marker of neoplastic progression in ulcerative colitis". Gastroenterology. 107 (2): 369–378. doi:10.1016/0016-5085(94)90161-9. PMID 8039614.
- Tsao JL, Yatabe Y, Salovaara R, Järvinen HJ, Mecklin JP, Aaltonen LA, et al. (February 2000). "Genetic reconstruction of individual colorectal tumor histories". Proceedings of the National Academy of Sciences of the United States of America. 97 (3): 1236–1241. Bibcode:2000PNAS...97.1236T. doi:10.1073/pnas.97.3.1236. PMC 15581. PMID 10655514.
- González-García I, Solé RV, Costa J (October 2002). "Metapopulation dynamics and spatial heterogeneity in cancer". Proceedings of the National Academy of Sciences of the United States of America. 99 (20): 13085–13089. Bibcode:2002PNAS...9913085G. doi:10.1073/pnas.202139299. PMC 130590. PMID 12351679.
- Harada T, Okita K, Shiraishi K, Kusano N, Kondoh S, Sasaki K (February 2002). "Interglandular cytogenetic heterogeneity detected by comparative genomic hybridization in pancreatic cancer". Cancer Research. 62 (3): 835–839. PMID 11830540.
- Murphy DS, Hoare SF, Going JJ, Mallon EE, George WD, Kaye SB, et al. (November 1995). "Characterization of extensive genetic alterations in ductal carcinoma in situ by fluorescence in situ hybridization and molecular analysis". Journal of the National Cancer Institute. 87 (22): 1694–1704. doi:10.1093/jnci/87.22.1694. PMID 7473818.
- Castro MA, Onsten TT, de Almeida RM, Moreira JC (June 2005). "Profiling cytogenetic diversity with entropy-based karyotypic analysis". Journal of Theoretical Biology. 234 (4): 487–495. Bibcode:2005JThBi.234..487C. doi:10.1016/j.jtbi.2004.12.006. PMID 15808870.
- Barrett MT, Sanchez CA, Prevo LJ, Wong DJ, Galipeau PC, Paulson TG, et al. (May 1999). "Evolution of neoplastic cell lineages in Barrett oesophagus". Nature Genetics. 22 (1): 106–109. doi:10.1038/8816. PMC 1559997. PMID 10319873.
- Hu W, Feng Z, Ma L, Wagner J, Rice JJ, Stolovitzky G, Levine AJ (March 2007). "A single nucleotide polymorphism in the MDM2 gene disrupts the oscillation of p53 and MDM2 levels in cells". Cancer Research. 67 (6): 2757–2765. doi:10.1158/0008-5472.CAN-06-2656. PMID 17363597.
- Goel A, Arnold CN, Niedzwiecki D, Carethers JM, Dowell JM, Wasserman L, et al. (May 2004). "Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers". Cancer Research. 64 (9): 3014–3021. doi:10.1158/0008-5472.CAN-2401-2. PMID 15126336.
- Kallioniemi A (February 2008). "CGH microarrays and cancer". Current Opinion in Biotechnology. 19 (1): 36–40. doi:10.1016/j.copbio.2007.11.004. PMID 18162393.
- Duesberg P, Rausch C, Rasnick D, Hehlmann R (November 1998). "Genetic instability of cancer cells is proportional to their degree of aneuploidy". Proceedings of the National Academy of Sciences of the United States of America. 95 (23): 13692–13697. Bibcode:1998PNAS...9513692D. doi:10.1073/pnas.95.23.13692. PMC 24881. PMID 9811862.
- Heng HH, Stevens JB, Liu G, Bremer SW, Ye KJ, Reddy PV, et al. (August 2006). "Stochastic cancer progression driven by non-clonal chromosome aberrations". Journal of Cellular Physiology. 208 (2): 461–472. doi:10.1002/jcp.20685. PMID 16688757. S2CID 33441988.
- Heng HH, Bremer SW, Stevens J, Ye KJ, Miller F, Liu G, Ye CJ (August 2006). "Cancer progression by non-clonal chromosome aberrations". Journal of Cellular Biochemistry. 98 (6): 1424–1435. doi:10.1002/jcb.20964. PMID 16676347. S2CID 23123441.
- Ye CJ, Liu G, Bremer SW, Heng HH (2007). "The dynamics of cancer chromosomes and genomes". Cytogenetic and Genome Research. 118 (2–4): 237–246. doi:10.1159/000108306. PMID 18000376. S2CID 22867025.
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. (February 2001). "Initial sequencing and analysis of the human genome". Nature. 409 (6822): 860–921. Bibcode:2001Natur.409..860L. doi:10.1038/35057062. PMID 11237011.
- Yost SE, Smith EN, Schwab RB, Bao L, Jung H, Wang X, et al. (August 2012). "Identification of high-confidence somatic mutations in whole genome sequence of formalin-fixed breast cancer specimens". Nucleic Acids Research. 40 (14): e107. doi:10.1093/nar/gks299. PMC 3413110. PMID 22492626.
- Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, et al. (May 2012). "Melanoma genome sequencing reveals frequent PREX2 mutations". Nature. 485 (7399): 502–506. Bibcode:2012Natur.485..502B. doi:10.1038/nature11071. PMC 3367798. PMID 22622578.
- Lee W, Jiang Z, Liu J, Haverty PM, Guan Y, Stinson J, et al. (May 2010). "The mutation spectrum revealed by paired genome sequences from a lung cancer patient". Nature. 465 (7297): 473–477. Bibcode:2010Natur.465..473L. doi:10.1038/nature09004. PMID 20505728. S2CID 4354035.
- Heng HH (August 2007). "Cancer genome sequencing: the challenges ahead". BioEssays. 29 (8): 783–794. doi:10.1002/bies.20610. PMID 17621658.
- Bielas JH, Loeb KR, Rubin BP, True LD, Loeb LA (November 2006). "Human cancers express a mutator phenotype". Proceedings of the National Academy of Sciences of the United States of America. 103 (48): 18238–18242. doi:10.1073/pnas.0607057103. PMC 1636340. PMID 17108085.
- Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, et al. (November 2007). "The genomic landscapes of human breast and colorectal cancers". Science. 318 (5853): 1108–1113. Bibcode:2007Sci...318.1108W. CiteSeerX 10.1.1.218.5477. doi:10.1126/science.1145720. PMID 17932254. S2CID 7586573.
- Halford S, Rowan A, Sawyer E, Talbot I, Tomlinson I (June 2005). "O(6)-methylguanine methyltransferase in colorectal cancers: detection of mutations, loss of expression, and weak association with G:C>A:T transitions". Gut. 54 (6): 797–802. doi:10.1136/gut.2004.059535. PMC 1774551. PMID 15888787.
- Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JO, et al. (May 2005). "Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer". Gastroenterology. 128 (5): 1160–1171. doi:10.1053/j.gastro.2005.01.056. PMID 15887099.
- Narayanan L, Fritzell JA, Baker SM, Liskay RM, Glazer PM (April 1997). "Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2". Proceedings of the National Academy of Sciences of the United States of America. 94 (7): 3122–3127. Bibcode:1997PNAS...94.3122N. doi:10.1073/pnas.94.7.3122. PMC 20332. PMID 9096356.
- Hegan DC, Narayanan L, Jirik FR, Edelmann W, Liskay RM, Glazer PM (December 2006). "Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6". Carcinogenesis. 27 (12): 2402–2408. doi:10.1093/carcin/bgl079. PMC 2612936. PMID 16728433.
- Tutt AN, van Oostrom CT, Ross GM, van Steeg H, Ashworth A (March 2002). "Disruption of Brca2 increases the spontaneous mutation rate in vivo: synergism with ionizing radiation". EMBO Reports. 3 (3): 255–260. doi:10.1093/embo-reports/kvf037. PMC 1084010. PMID 11850397.
- Goel A, Boland CR (December 2012). "Epigenetics of colorectal cancer". Gastroenterology. 143 (6): 1442–1460.e1. doi:10.1053/j.gastro.2012.09.032. PMC 3611241. PMID 23000599.
- Schnekenburger M, Diederich M (March 2012). "Epigenetics Offer New Horizons for Colorectal Cancer Prevention". Current Colorectal Cancer Reports. 8 (1): 66–81. doi:10.1007/s11888-011-0116-z. PMC 3277709. PMID 22389639.
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW (March 2013). "Cancer genome landscapes". Science. 339 (6127): 1546–1558. Bibcode:2013Sci...339.1546V. doi:10.1126/science.1235122. PMC 3749880. PMID 23539594.
- Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr AR, James KD, Turner DJ, et al. (September 2010). "Orphan CpG islands identify numerous conserved promoters in the mammalian genome". PLOS Genetics. 6 (9): e1001134. doi:10.1371/journal.pgen.1001134. PMC 2944787. PMID 20885785.
- Wei J, Li G, Dang S, Zhou Y, Zeng K, Liu M (2016). "Discovery and Validation of Hypermethylated Markers for Colorectal Cancer". Disease Markers. 2016: 2192853. doi:10.1155/2016/2192853. PMC 4963574. PMID 27493446.
- Beggs AD, Jones A, El-Bahrawy M, El-Bahwary M, Abulafi M, Hodgson SV, Tomlinson IP (April 2013). "Whole-genome methylation analysis of benign and malignant colorectal tumours". The Journal of Pathology. 229 (5): 697–704. doi:10.1002/path.4132. PMC 3619233. PMID 23096130.
- Bird A (January 2002). "DNA methylation patterns and epigenetic memory". Genes & Development. 16 (1): 6–21. doi:10.1101/gad.947102. PMID 11782440.
- Czerniak B, Chaturvedi V, Li L, Hodges S, Johnston D, Roy JY, et al. (February 1999). "Superimposed histologic and genetic mapping of chromosome 9 in progression of human urinary bladder neoplasia: implications for a genetic model of multistep urothelial carcinogenesis and early detection of urinary bladder cancer". Oncogene. 18 (5): 1185–1196. doi:10.1038/sj.onc.1202385. PMID 10022124.
- Majewski T, Lee S, Jeong J, Yoon DS, Kram A, Kim MS, et al. (July 2008). "Understanding the development of human bladder cancer by using a whole-organ genomic mapping strategy". Laboratory Investigation; A Journal of Technical Methods and Pathology. 88 (7): 694–721. doi:10.1038/labinvest.2008.27. PMC 2849658. PMID 18458673.
- Zhang W, Hanks AN, Boucher K, Florell SR, Allen SM, Alexander A, et al. (January 2005). "UVB-induced apoptosis drives clonal expansion during skin tumor development". Carcinogenesis. 26 (1): 249–257. doi:10.1093/carcin/bgh300. PMC 2292404. PMID 15498793.
- Sidransky D, Mikkelsen T, Schwechheimer K, Rosenblum ML, Cavanee W, Vogelstein B (February 1992). "Clonal expansion of p53 mutant cells is associated with brain tumour progression". Nature. 355 (6363): 846–847. Bibcode:1992Natur.355..846S. doi:10.1038/355846a0. PMID 1311419. S2CID 4318673.
- Bardeesy N, Beckwith JB, Pelletier J (January 1995). "Clonal expansion and attenuated apoptosis in Wilms' tumors are associated with p53 gene mutations". Cancer Research. 55 (2): 215–219. PMID 7812946.
- McDonald SA, Greaves LC, Gutierrez-Gonzalez L, Rodriguez-Justo M, Deheragoda M, Leedham SJ, et al. (February 2008). "Mechanisms of field cancerization in the human stomach: the expansion and spread of mutated gastric stem cells". Gastroenterology. 134 (2): 500–510. doi:10.1053/j.gastro.2007.11.035. PMID 18242216.
- Lee S, Jeong J, Majewski T, Scherer SE, Kim MS, Tuziak T, et al. (August 2007). "Forerunner genes contiguous to RB1 contribute to the development of in situ neoplasia". Proceedings of the National Academy of Sciences of the United States of America. 104 (34): 13732–13737. Bibcode:2007PNAS..10413732L. doi:10.1073/pnas.0701771104. PMC 1949496. PMID 17702869.
- McDonald SA, Preston SL, Greaves LC, Leedham SJ, Lovell MA, Jankowski JA, et al. (April 2006). "Clonal expansion in the human gut: mitochondrial DNA mutations show us the way". Cell Cycle. 5 (8): 808–811. doi:10.4161/cc.5.8.2641. PMID 16628008.
- Park IW, Wistuba II, Maitra A, Milchgrub S, Virmani AK, Minna JD, Gazdar AF (November 1999). "Multiple clonal abnormalities in the bronchial epithelium of patients with lung cancer". Journal of the National Cancer Institute. 91 (21): 1863–1868. doi:10.1093/jnci/91.21.1863. PMID 10547393.
- Tiu R, Gondek L, O'Keefe C, Maciejewski JP (August 2007). "Clonality of the stem cell compartment during evolution of myelodysplastic syndromes and other bone marrow failure syndromes". Leukemia. 21 (8): 1648–1657. doi:10.1038/sj.leu.2404757. PMID 17554386.
- Mehra R, Tomlins SA, Yu J, Cao X, Wang L, Menon A, et al. (May 2008). "Characterization of TMPRSS2-ETS gene aberrations in androgen-independent metastatic prostate cancer". Cancer Research. 68 (10): 3584–3590. doi:10.1158/0008-5472.CAN-07-6154. PMC 2677168. PMID 18483239.
- Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Blount PL, Reid BJ (October 2004). "The combination of genetic instability and clonal expansion predicts progression to esophageal adenocarcinoma". Cancer Research. 64 (20): 7629–7633. doi:10.1158/0008-5472.CAN-04-1738. PMID 15492292.
- Beerenwinkel N, Antal T, Dingli D, Traulsen A, Kinzler KW, Velculescu VE, et al. (November 2007). "Genetic progression and the waiting time to cancer". PLOS Computational Biology. 3 (11): e225. arXiv:0707.3770. Bibcode:2007PLSCB...3..225B. doi:10.1371/journal.pcbi.0030225. PMC 2065895. PMID 17997597.
- Slaughter DP, Southwick HW, Smejkal W (September 1953). "Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin". Cancer. 6 (5): 963–968. doi:10.1002/1097-0142(195309)6:5<963::AID-CNCR2820060515>3.0.CO;2-Q. PMID 13094644. S2CID 6736946.
- Bernstein C, Bernstein H, Payne CM, Dvorak K, Garewal H (February 2008). "Field defects in progression to gastrointestinal tract cancers". Cancer Letters. 260 (1–2): 1–10. doi:10.1016/j.canlet.2007.11.027. PMC 2744582. PMID 18164807.
- Louhelainen J, Wijkström H, Hemminki K (July 2000). "Initiation-development modelling of allelic losses on chromosome 9 in multifocal bladder cancer". European Journal of Cancer. 36 (11): 1441–1451. doi:10.1016/S0959-8049(00)00127-1. PMID 10899659.
- Desper R, Jiang F, Kallioniemi OP, Moch H, Papadimitriou CH, Schäffer AA (1999). "Inferring tree models for oncogenesis from comparative genome hybridization data". Journal of Computational Biology. 6 (1): 37–51. CiteSeerX 10.1.1.53.9617. doi:10.1089/cmb.1999.6.37. PMID 10223663.
- Bast, F. 2012. Cancer Phylogenetics: Computational Modeling of Tumor Evolution. In R. Tuteja (Ed.), Bioinformatics: Genome Bioinformatics and Computational Biology (pp. 211-230).Nova Publishers New York. 211-230
- Wright S (March 1931). "Evolution in Mendelian Populations". Genetics. 16 (2): 97–159. doi:10.1093/genetics/16.2.97. PMC 1201091. PMID 17246615.
- Wright S. Evolution and genetics of populations. Vol. 2, University of Chicago Press (1969)
- Nowak MA, Sigmund K (February 2004). "Evolutionary dynamics of biological games" (PDF). Science. 303 (5659): 793–799. Bibcode:2004Sci...303..793N. doi:10.1126/science.1093411. PMID 14764867. S2CID 2966169.
- Vincent T. L. and Brown J. S. Evolutionary game theory, natural selection, and Darwinian dynamics. Cambridge University Press 2005
- Vincent TL, Gatenby RA (April 2008). "An evolutionary model for initiation, promotion, and progression in carcinogenesis". International Journal of Oncology. 32 (4): 729–737. doi:10.3892/ijo.32.4.729. PMID 18360700.
- Maley CC, Reid BJ, Forrest S (August 2004). "Cancer prevention strategies that address the evolutionary dynamics of neoplastic cells: simulating benign cell boosters and selection for chemosensitivity". Cancer Epidemiology, Biomarkers & Prevention. 13 (8): 1375–1384. doi:10.1158/1055-9965.1375.13.8. PMID 15298961. S2CID 1143689.
- Spencer SL, Gerety RA, Pienta KJ, Forrest S (August 2006). "Modeling somatic evolution in tumorigenesis". PLOS Computational Biology. 2 (8): e108. Bibcode:2006PLSCB...2..108S. doi:10.1371/journal.pcbi.0020108. PMC 1550273. PMID 16933983.
- Axelrod R, Axelrod DE, Pienta KJ (September 2006). "Evolution of cooperation among tumor cells". Proceedings of the National Academy of Sciences of the United States of America. 103 (36): 13474–13479. doi:10.1073/pnas.0606053103. PMC 1557388. PMID 16938860.
- Shackleton M, Quintana E, Fearon ER, Morrison SJ (September 2009). "Heterogeneity in cancer: cancer stem cells versus clonal evolution". Cell. 138 (5): 822–829. doi:10.1016/j.cell.2009.08.017. PMID 19737509. S2CID 2615068.
- Bapat SA (June 2007). "Evolution of cancer stem cells". Seminars in Cancer Biology. 17 (3): 204–213. doi:10.1016/j.semcancer.2006.05.001. PMID 16787749.
- Dalerba P, Cho RW, Clarke MF (2007). "Cancer stem cells: models and concepts". Annual Review of Medicine. 58: 267–284. doi:10.1146/annurev.med.58.062105.204854. PMID 17002552.
- Chabner BA, Roberts TG (January 2005). "Timeline: Chemotherapy and the war on cancer". Nature Reviews. Cancer. 5 (1): 65–72. doi:10.1038/nrc1529. PMID 15630416. S2CID 205467419.
- Schimke RT (May 1984). "Gene amplification, drug resistance, and cancer". Cancer Research. 44 (5): 1735–1742. PMID 6713376.
- Curt GA, Carney DN, Cowan KH, Jolivet J, Bailey BD, Drake JC, et al. (January 1983). "Unstable methotrexate resistance in human small-cell carcinoma associated with double minute chromosomes". The New England Journal of Medicine. 308 (4): 199–202. doi:10.1056/NEJM198301273080406. PMID 6294518. S2CID 44868799.
- Carman MD, Schornagel JH, Rivest RS, Srimatkandada S, Portlock CS, Duffy T, Bertino JR (January 1984). "Resistance to methotrexate due to gene amplification in a patient with acute leukemia". Journal of Clinical Oncology. 2 (1): 16–20. doi:10.1200/JCO.1984.2.1.16. PMID 6583326.
- Horns RC, Dower WJ, Schimke RT (January 1984). "Gene amplification in a leukemic patient treated with methotrexate". Journal of Clinical Oncology. 2 (1): 2–7. doi:10.1200/JCO.1984.2.1.2. PMID 6583327.
- Trent JM, Buick RN, Olson S, Horns RC, Schimke RT (January 1984). "Cytologic evidence for gene amplification in methotrexate-resistant cells obtained from a patient with ovarian adenocarcinoma". Journal of Clinical Oncology. 2 (1): 8–15. doi:10.1200/JCO.1984.2.1.8. PMID 6699660.
- Wang TL, Diaz LA, Romans K, Bardelli A, Saha S, Galizia G, et al. (March 2004). "Digital karyotyping identifies thymidylate synthase amplification as a mechanism of resistance to 5-fluorouracil in metastatic colorectal cancer patients". Proceedings of the National Academy of Sciences of the United States of America. 101 (9): 3089–3094. Bibcode:2004PNAS..101.3089W. doi:10.1073/pnas.0308716101. PMC 420348. PMID 14970324.
- Gorre ME, Sawyers CL (July 2002). "Molecular mechanisms of resistance to STI571 in chronic myeloid leukemia". Current Opinion in Hematology. 9 (4): 303–307. doi:10.1097/00062752-200207000-00007. PMID 12042704. S2CID 34233816.
- Roche-Lestienne C, Preudhomme C (April 2003). "Mutations in the ABL kinase domain pre-exist the onset of imatinib treatment". Seminars in Hematology. 40 (2 Suppl 2): 80–82. doi:10.1053/shem.2003.50046. PMID 12783380.
- Shah NP, Skaggs BJ, Branford S, Hughes TP, Nicoll JM, Paquette RL, Sawyers CL (September 2007). "Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency". The Journal of Clinical Investigation. 117 (9): 2562–2569. doi:10.1172/JCI30890. PMC 1940237. PMID 17710227.
- Tamborini E, Bonadiman L, Greco A, Albertini V, Negri T, Gronchi A, et al. (July 2004). "A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient". Gastroenterology. 127 (1): 294–299. doi:10.1053/j.gastro.2004.02.021. PMID 15236194.
- Chen LL, Trent JC, Wu EF, Fuller GN, Ramdas L, Zhang W, et al. (September 2004). "A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors". Cancer Research. 64 (17): 5913–5919. doi:10.1158/0008-5472.CAN-04-0085. PMID 15342366.
- Engelman JA, Jänne PA (May 2008). "Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer". Clinical Cancer Research. 14 (10): 2895–2899. doi:10.1158/1078-0432.CCR-07-2248. PMID 18483355.
- Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, et al. (February 2005). "EGFR mutation and resistance of non-small-cell lung cancer to gefitinib". The New England Journal of Medicine. 352 (8): 786–792. doi:10.1056/NEJMoa044238. PMID 15728811.
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. (May 2007). "MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling". Science. 316 (5827): 1039–1043. Bibcode:2007Sci...316.1039E. doi:10.1126/science.1141478. PMID 17463250. S2CID 23254145.
- Ring A, Dowsett M (December 2004). "Mechanisms of tamoxifen resistance". Endocrine-Related Cancer. 11 (4): 643–658. doi:10.1677/erc.1.00776. PMID 15613444.
- Osborne CK (November 1998). "Tamoxifen in the treatment of breast cancer". The New England Journal of Medicine. 339 (22): 1609–1618. doi:10.1056/NEJM199811263392207. PMID 9828250.
- Encarnación CA, Ciocca DR, McGuire WL, Clark GM, Fuqua SA, Osborne CK (1993). "Measurement of steroid hormone receptors in breast cancer patients on tamoxifen". Breast Cancer Research and Treatment. 26 (3): 237–246. doi:10.1007/BF00665801. PMID 8251648. S2CID 9716966.
- Johnston SR, Saccani-Jotti G, Smith IE, Salter J, Newby J, Coppen M, et al. (August 1995). "Changes in estrogen receptor, progesterone receptor, and pS2 expression in tamoxifen-resistant human breast cancer". Cancer Research. 55 (15): 3331–3338. PMID 7614468.
- Jordan VC, O'Malley BW (December 2007). "Selective estrogen-receptor modulators and antihormonal resistance in breast cancer". Journal of Clinical Oncology. 25 (36): 5815–5824. doi:10.1200/JCO.2007.11.3886. PMID 17893378.
- Beverage JN, Sissung TM, Sion AM, Danesi R, Figg WD (September 2007). "CYP2D6 polymorphisms and the impact on tamoxifen therapy". Journal of Pharmaceutical Sciences. 96 (9): 2224–2231. doi:10.1002/jps.20892. PMID 17518364.
- Taplin ME, Bubley GJ, Ko YJ, Small EJ, Upton M, Rajeshkumar B, Balk SP (June 1999). "Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist". Cancer Research. 59 (11): 2511–2515. PMID 10363963.
- Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinänen R, Palmberg C, et al. (April 1995). "In vivo amplification of the androgen receptor gene and progression of human prostate cancer". Nature Genetics. 9 (4): 401–406. doi:10.1038/ng0495-401. PMID 7795646. S2CID 20120114.
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. (December 2006). "Glioma stem cells promote radioresistance by preferential activation of the DNA damage response". Nature. 444 (7120): 756–760. Bibcode:2006Natur.444..756B. doi:10.1038/nature05236. PMID 17051156. S2CID 4340708.
- Kim Y, Kim KH, Lee J, Lee YA, Kim M, Lee SJ, et al. (March 2012). "Wnt activation is implicated in glioblastoma radioresistance". Laboratory Investigation; A Journal of Technical Methods and Pathology. 92 (3): 466–473. doi:10.1038/labinvest.2011.161. PMID 22083670.
- Pepper JW, Scott Findlay C, Kassen R, Spencer SL, Maley CC (February 2009). "Cancer research meets evolutionary biology". Evolutionary Applications. 2 (1): 62–70. doi:10.1111/j.1752-4571.2008.00063.x. PMC 3352411. PMID 25567847.
- Jain RK, Forbes NS (December 2001). "Can engineered bacteria help control cancer?". Proceedings of the National Academy of Sciences of the United States of America. 98 (26): 14748–14750. Bibcode:2001PNAS...9814748J. doi:10.1073/pnas.261606598. PMC 64926. PMID 11752416.