Familial dysautonomia
Familial dysautonomia (FD), also known as Riley-Day Syndrome, is a rare,[2] progressive,[3] recessive genetic disorder of the autonomic nervous system[2] that affects the development and survival of sensory, sympathetic and some parasympathetic neurons in the autonomic and sensory nervous system.
| Familial dysautonomia | |
|---|---|
| Other names | Riley–Day syndrome[1] and Hereditary sensory and autonomic neuropathy type III (HSAN-III) |
 | |
| Facial features of a patient with familial dysautonomia over time. Note flattening of upper lip. By age 10 years prominence of lower jaw is apparent and by age 19 years there is mild erosion of right nostril due to inadvertent self-mutilation. | |
| Specialty | Neurology |
FD results in variable symptoms, including insensitivity to pain, inability to produce tears, poor growth and labile blood pressure (episodic hypertension and postural hypotension). People with FD have frequent vomiting crises, pneumonia, problems with speech and movement, difficulty swallowing, inappropriate perception of heat, pain and taste as well as unstable blood pressure and gastrointestinal dysmotility.
Originally reported by Drs. Conrad Milton Riley and Richard Lawrence Day in 1949,[4] FD is one example of a group of disorders known as hereditary sensory and autonomic neuropathies (HSAN).[5] All HSAN are characterized by widespread sensory dysfunction and variable autonomic dysfunction caused by incomplete development of sensory and autonomic neurons. The disorders are believed to be genetically distinct from each other.
Signs and symptoms
Signs and symptoms of familial dysautonomia usually commence during infancy and worsen with age, and may include: gastrointestinal dysmotility (including erratic gastric emptying, gastroesophageal reflux, abnormal esophageal peristalsis, oropharyngeal incoordination),[3] dysphagia (as poor suckling in infancy) and frequent choking/gagging, recurrent vomiting, poor weight gain[6]/growth,[7] delayed development (especially walking) and puberty (especially in girls), recurrent aspiration pneumonia (due to inhalation of food or vomitus)[6] with possible secondary chronic lung disease,[3] absence of overflow tears during crying, corneal ulcers, red skin blotches and excessive sweating (often during eating or excitement), breath-holding spells, slurred speech/nasal voice, tongue ulcers (from accidental self-injuries), hyporeflexia (variable absence of deep tendon reflexes[8]), hypotonia, enuresis, arrhythmias, hypertension (including episodic hypertension in response to emotional stress or visceral pain[3]), hypotension (including orthostatic hypertension[7] with compensatory tachycardia (invariably present)[3]),[6] impaired (but not absent[3]) temperature and pain perception (leading to frequent accidental injury),[2][6] impaired proprioception, a smooth glossy tongue,[6] scoliosis (with possibly secondary restrictive lung disease[3]),[7] abnormal gait,[9] short stature, chronic renal failure (common), visual impairment, variable cognitive ability, characteristic facial features that develop with time, impaired vibration perception, lack of fungiform papilla of the tongue,[3] and impaired taste perception (especially for sweetness).[8]
- Autonomic crises - In children with FD, recurrent episodes of vomiting may occur. Such episodes may be triggered by physical (e.g. infection) or emotional stress, may occur every 15–20 minutes for over 24 hours, and may be accompanied with: significant hypertension, drenching sweat, breathing issues,[6] fever, tachycardia, aspiration pneumonia,[8] skin blotches, drooling, and negative personality change.
- Pain insensitivity - Insensitivity or indifference to painful stimuli may lead to frequent/progressive self-mutilation, burns, and ulcers. There may be self-mutilation of the tongue (especially in toddlers during teething), lips, and cheeks, or loss of teeth. Compulsive oral biting may result in ulcers, or tumour-like masses (Riga-Fede disease).[8]
Progression
Familial dysautonomia presents with progressive[3] age-specific symptoms.
Though usually not diagnosed until several years of age, generalised signs of FD are present in during the newborn period >80% of those affected.[8]Dysmorpnic facial features are not directly inherent to the disorder, however, facial asymmetry and a straightened mouth eventually develop due to abnormal tone and molding of facial bones.[8][3]
Perinatal
A very high incidence of breech presentation has been noted among infants with FD.[8][10][11] A lower birth weight as compared to siblings,[10] premature birth, and intrauterine growth restriction[8] have also been noted.
Neonatal
During the neonatal period, there may be hypotonia, respiratory insufficiency, poor feeding with difficulty swallowing and aspiration, developmental delay, short stature, scoliosis, and corneal disease.[8]
Infancy
Issues related to the disorder first appear during infancy. Early manifestations include hypotonia, feeding difficulty (impaired swallowing and suckling[2]), poor growth, absence of tears, frequent lung infections, and poor body temperature control (infants may display cold hands and feet[2]). Developmental milestones (e.g. walking, speech) may or may not be delayed.[7]
In infants with FD, a lack of overflow tears during emotional crying may be noted after the age of 7 months (until this age, overflow emotional tearing may also not occur in unaffected infants;[12] overflow tearing is absent in neonates and begins to appear only after 2–3 months of age[8]).[12]
Affected infants' hands may alternatively appear cool and mottled (from vasoconstriction), or red and swollen (from vasodilation).[8] Red skin blotching is often precipitated by emotional excitement.[8]
In older infants and young children, breath-holding spells may occur, possibly leading to cyanosis or fainting. Breath-holding behaviour usually ceases by age 6.[7]
Children
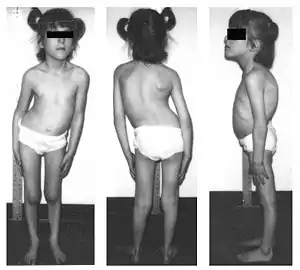
Breath-holding behaviour usually resolves by age 6. In school-age children, there may be bed wetting, vomiting episodes, impaired pain and temperature perception, impaired blood pressure control (including orthostatic hypotension, an hypertension during periods of psychological excitement or vomiting), learning disabilities (e.g. short attention span; learning disabilities are present in about a third of those with FD, and may require special education), scoliosis, poor bone quality and bone fractures, and kidney and heart issues.[7]
Adolescence and adulthood
Issues that tend to commence during adolescence or early adulthood include lung damage due to multiple respiratory infections, impaired kidney function, and impaired vision (due to atrophy of the optic nerve).[7] By adulthood, there are often mounting difficulties with balance and unaided walking.[7]
Cause
Familial dysautonomia is the result of mutations in IKBKAP gene on chromosome 9, which encodes for the IKAP protein (IkB kinase complex-associated protein). There have been three mutations in IKBKAP identified in individuals with FD. The most common FD-causing mutation occurs in intron 20 of the donor gene. Conversion of T→C in intron 20 of the donor gene resulted in shift splicing that generates an IKAP transcript lacking exon 20. Translation of this mRNA results in a truncated protein lacking all of the amino acids encoded by exons 20–37. Another less common mutation is a G→C conversion resulting in one amino acid mutation in 696, where Proline substitutes normal Arginine. The decreased amount of functional IKAP protein in cells causes familial dysautonomia.
Diagnosis
Clinical diagnosis
A clinical diagnosis of FD is supported by a constellation of criteria:
- No fungiform papillae on the tongue
- Decreased deep tendon reflexes
- Lack of an axon flare following intradermal histamine
- No overflow tears with emotional crying
Genetic testing
Genetic testing is performed on a small sample of blood from the tested individual. The DNA is examined with a designed probe specific to the known mutations. The accuracy of the test is above 99%. Dr. Anat Blumenfeld of the Hadassah Medical center in Jerusalem identified chromosome number 9 as the responsible chromosome.
Prenatal testing
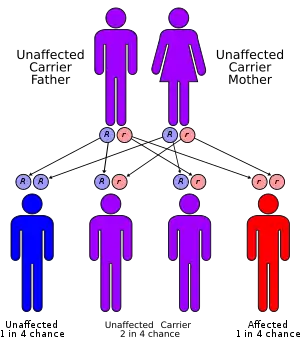
Familial dysautonomia is inherited in an autosomal recessive pattern, which means two copies of the gene in each cell are altered. If both parents are shown to be carriers by genetic testing, there is a 25% chance that the child will have FD. For pregnancies at increased risk for FD, preimplantation genetic diagnosis or prenatal diagnosis by amniocentesis (at 15–17 weeks) or chorionic villus sampling (at 10–14 weeks) is possible.
Management
There is currently no cure for FD. There are only two treatment centers, one at New York University Hospital[13] and one at the Sheba Medical Center in Israel.[14] One is being planned for the San Francisco area.[15] Although the FD-causing gene has been identified and it seems to have tissue specific expression, there is no definitive treatment at present.
A major issue has been aspiration pneumonia. Fundoplications (by preventing regurgitation) and gastrostomy tubes (to provide non-oral nutrition) have reduced the frequency of hospitalization. Other issues which can be treated include FD crises, scoliosis, and various eye conditions due to limited or no tears.
Treatment of FD remains preventative, symptomatic and supportive. FD does not express itself in a consistent manner. The type and severity of symptoms displayed vary among patients and even at different ages on the same patients so patients should have specialized individual treatment plans. Medications are used to control vomiting, eye dryness, and abnormal blood pressure. Common management strategies include: artificial tears, appropriate feeding strategy (maintenance of adequate nutrition, avoidance of aspiration (thickened formula and different shaped nipples for infants)), daily chest physiotherapy (nebulization, bronchodilators, and postural drainage) for chronic pulmonary disease, pharmaceutical management of autonomic features (e.g. intravenous or rectal diazepam, or rectal chloral hydrate), preventing accidental injury, prevention of orthostatic hypotension (hydration, leg exercise, frequent small meals, a high-salt diet, and medication (e.g. with fludrocortisone)), treatment of orthopedic problems, compensating labile blood pressure.
Parents and patients should be informed regarding daily eye care and early signs of corneal problems, as well as punctal cautery. Informing patients and caretakers has resulted in decreased corneal scarring and need for more aggressive surgical measures such as tarsorrhaphy, conjunctival flaps, and corneal transplants.
Prognosis
Average age of death is in the 3rd decade of life, however, affected persons may live into the 7th decade of life.[3] Death occurs in 50% of the affected individuals by age 30. The outlook for patients with FD depends on the particular diagnostic category. Patients with chronic, progressive, generalized dysautonomia in the setting of central nervous system degeneration have a generally poor long-term prognosis. Death can occur from pneumonia, acute respiratory failure, or sudden cardiopulmonary arrest in such patients.
The survival rate and quality of life have increased since the mid-1980s mostly due to a greater understanding of the most dangerous symptoms. At present, FD patients can be expected to function independently if treatment is begun early and if major disabilities are avoided.
Epidemiology
Familial dysautonomia is seen almost exclusively in Ashkenazi Jews and is inherited in an autosomal recessive fashion. Both parents must be carriers in order for a child to be affected. The carrier frequency in Jews of Eastern and Central European (Ashkenazi) ancestry is about 1/30, while the carrier frequency in non-Jews is unknown. If both parents are carriers, there is a one in four, or 25%, chance with each pregnancy for an affected child. Genetic counseling and genetic testing is recommended for families who may be carriers of familial dysautonomia.
Worldwide, there have been approximately 600 diagnoses recorded since discovery of the disease, with approximately 350 of them still living.[16]
Research
In January 2001, researchers at Fordham University and Massachusetts General Hospital simultaneously reported finding the genetic mutation that causes FD, a discovery that opens the door to many diagnostic and treatment possibilities.[17][18] Genetic screening subsequently became available in 2001, enabling Ashkenazi Jews to find out if they are carriers.
Stem-cell therapy has been proposed as a potential future treatment. Eventually, treatment could be given in utero. Research into treatments is being funded by foundations organized and run by parents of those with FD. There is no governmental support beyond recognizing those diagnosed with FD as eligible for certain programs.[19]
References
- pediatriconcall.com Archived 2007-04-30 at the Wayback Machine
- "Dysautonomia, Familial". NORD (National Organization for Rare Disorders). Retrieved 2020-05-31.
- "Orphanet: Familial dysautonomia". www.orpha.net. Retrieved 2020-05-31.
- Riley CM, Day RL, Greely D, Langford WS (1949). "Central autonomic dysfunction with defective lacrimation". Pediatrics. 3 (4): 468–77. doi:10.1542/peds.3.4.468. PMID 18118947. S2CID 245200408.
- Axelrod FB (2002). "Hereditary sensory and autonomic neuropathies. Familial dysautonomia and other HSANs". Clin Auton Res. 12. Suppl 1 (7): I2–14. doi:10.1007/s102860200014. PMID 12102459. S2CID 44306353.
- Publishing, Harvard Health (21 December 2018). "Familial Dysautonomia". Harvard Health. Retrieved 2020-05-30.
- Reference, Genetics Home. "Familial dysautonomia". Genetics Home Reference. Retrieved 2020-05-30.
- Neonatal and infant dermatology. Eichenfield, Lawrence F.,, Frieden, Ilona J.,, Mathes, Erin F.,, Zaenglein, Andrea L. (Third ed.). London. 5 September 2014. ISBN 978-1-4557-2639-4. OCLC 885376300.
{{cite book}}: CS1 maint: others (link) - Portnoy, Sigal; Maayan, Channa; Tsenter, Jeanna; Ofran, Yonah; Goldman, Vladimir; Hiller, Nurit; Karniel, Naama; Schwartz, Isabella (2018-04-26). "Characteristics of ataxic gait in familial dysautonomia patients". PLOS ONE. 13 (4): e0196599. Bibcode:2018PLoSO..1396599P. doi:10.1371/journal.pone.0196599. ISSN 1932-6203. PMC 5919612. PMID 29698477.
- Axelrod, Felicia B.; Leistner, Hedi L.; Porges, Robert F. (1974-01-01). "Breech presentation among infants with familial dysautonomia". The Journal of Pediatrics. 84 (1): 107–109. doi:10.1016/S0022-3476(74)80564-0. ISSN 0022-3476. PMID 12119926.
- Mostello, D.; Chang, J. J.; Bai, F.; Wang, J.; Guild, C.; Stamps, K.; Leet, T. L. (January 2014). "Breech presentation at delivery: a marker for congenital anomaly?". Journal of Perinatology. 34 (1): 11–15. doi:10.1038/jp.2013.132. ISSN 1476-5543. PMID 24157495. S2CID 21579907.
- "Familial Dysautonomia Workup: Laboratory Studies". emedicine.medscape.com. Retrieved 2020-05-31.
- Dysautonomia Treatment and Evaluation Center Archived 2006-01-14 at the Wayback Machine
- Sheba Medical Center
- "San Francisco To Get a Genetics Center - Forward.com". 5 August 2005. Retrieved 2007-11-02.
- "FD History & Statistics - Dysautonomia Foundation - Familial Dysautonomia (FD)". Archived from the original on 2009-04-18. Retrieved 2009-04-02.
- Anderson SL, Coli R, Daly IW, Kichula EA, Rork MJ, Volpi SA, Ekstein J, Rubin BY (2001). "Familial dysautonomia is caused by mutations of the IKAP gene". Am J Hum Genet. 68 (3): 753–8. doi:10.1086/318808. PMC 1274486. PMID 11179021.
- Slaugenhaupt SA, Blumenfeld A, Gill SP, Leyne M, Mull J, Cuajungco MP, Liebert CB, Chadwick B, Idelson M, Reznik L, Robbins C, Makalowska I, Brownstein M, Krappmann D, Scheidereit C, Maayan C, Axelrod FB, Gusella JF (2001). "Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia". Am J Hum Genet. 68 (3): 598–605. doi:10.1086/318810. PMC 1274473. PMID 11179008.
- Benefits for people with FD Archived 2011-07-26 at the Wayback Machine – from Dysautonomia foundation
Further reading
- Axelrod FB, Hilz MJ (2003). "Inherited autonomic neuropathies". Semin Neurol. 23 (4): 381–90. doi:10.1055/s-2004-817722. PMID 15088259.
- Axelrod FB (2004). "Familial dysautonomia". Muscle Nerve. 29 (3): 352–63. doi:10.1002/mus.10499. PMID 14981733. S2CID 222032372.
- Slaugenhaupt SA, Gusella JF (2002). "Familial dysautonomia". Curr Opin Genet Dev. 12 (3): 307–11. doi:10.1016/S0959-437X(02)00303-9. PMID 12076674.
- Felicia B Axelrod; Gabrielle Gold-von Simson (October 3, 2007). "Hereditary sensory and autonomic neuropathies: types II, III, and IV". Orphanet Journal of Rare Diseases. 2 (39): 39. doi:10.1186/1750-1172-2-39. PMC 2098750. PMID 17915006.