18p-
18p- is a genetic condition caused by a deletion of all or part of the short arm (the p arm) of chromosome 18. It occurs in about 1 of every 50,000 births.[1]
| 18p- | |
|---|---|
| Other names | Partial monosomy 18p, de Grouchy syndrome type 1 |
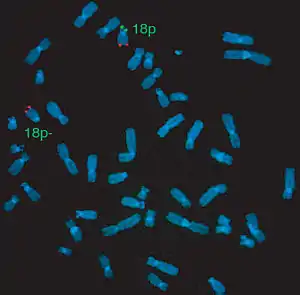 | |
| In situ hybridization. 18p (green) and 18q (red) with subtelomeric probes showing 18p deletion in the patient with De Grouchy syndrome type I (deletion 18p) | |
| Specialty | Medical genetics |
Signs and symptoms
.png.webp)
18p- causes a wide range of medical and developmental concerns. There is significant variation in severity. This variation is due to the variability of the deletion size and breakpoints.[2]
Congenital anomalies
About 10–15% of individuals with 18p- have holoprosencephaly.
Approximately 10% of people with 18p- have a congenital heart anomaly. There does not appear to be a specific type of heart defect associated with a deletion of the short arm of chromosome 18. Septal defects, tetralogy of Fallot, dextrocardia, and coarctation of the aorta have all been reported in infants with 18p-.
Neurologic
Hypotonia is frequently seen in the 18p- population. Seizures, though uncommon, have been reported in people with 18p-. Dystonia has also been diagnosed in a small minority of young adults with 18p-. Also, tethered cord has been reported in a few people with 18p-.
Vision
Ptosis is quite common among people with 18p-. In many cases, surgical correction is required. Refractive errors, such as myopia, hyperopia, and astigmatism, are also prevalent. Strabismus has been reported in infants and children with 18p-. Nystagmus is also present in a minority of individuals.
Ear and Sinus Infections
Children with 18p- have an increased incidence of ear infections, often requiring the placement of PE tubes.
Hearing
Conductive hearing loss may occur due to otitis media.
Gastrointestinal
Chronic constipation is a frequent complaint in the 18p- population. Other abdominal abnormalities that have been reported include inguinal hernias; malrotation of the gut; and abnormalities of the spleen.
Genitourinary
Genitourinary abnormalities are not common in 18p-. There have been a few cases of small penis and cryptorchidism in males and uterine abnormalities in females.
Orthopedics
There have been several orthopedic concerns identified in individuals with 18p-. These include pes planus, clubfoot, scoliosis and/or kyphosis, pectus abnormalities, cubitus valgus, congenital hip dysplasia, spina bifida occulta, and genu valgum.
Endocrinology
Growth hormone deficiency has been reported in several individuals with 18p-, though not at the same frequency as in the distal 18q- population. Panhypopituitarism and hypothyroidism have each been diagnosed in a handful of individuals. Also, ketotic hypoglycemia has been reported in several individuals and usually presents itself around the age of three.
Psychiatry
There is an increased incidence of psychiatric conditions within the distal 18p- population. In one study, 2 of 3 people with 18p- had an anxiety disorder, 1 of 3 had a communication disorder, and 1 of 3 had a motor skills disorders, and 1 of 3 had a stereotypic movement disorder. Additional research with a larger number of subjects is necessary to confirm these findings.
Cognition and adaptive skills
Cognitive ability in individuals with 18p- varies widely, with most falling in the mild to moderate range of impairment, though there have been some reports of people with impairment in the severe to profound range. These individuals may have had holoprosencephaly, which is frequently associated with severe impairment.
Speech deficits are quite common within this population. Frequently, expressive speech lags behind other developmental parameters.
Dysmorphology
Common facial features include a flat and broad nasal bridge; epicanthic folds; wide mouth; short philtrum; everted lower lip; small and slightly receding chin during childhood. The ears may be low-set and posteriorly rotated. The posterior hairline may be low.
Genetics
18p- describes a deletion of the short arm of chromosome 18.[1] About half of the people with deletions have a breakpoint at the centromere. Those with it are said to have centromeric 18p-, and those without are said to have non-centromeric 18p-.
Diagnosis
Suspicion of a chromosome abnormality is typically raised due to the presence of developmental delays or birth defects. Diagnosis of 18p- is usually made via a blood sample. A routine chromosome analysis, or karyotype, is usually used to make the initial diagnosis, although it may also be made by microarray analysis. Increasingly, microarray analysis is also being used to clarify breakpoints. Prenatal diagnosis is possible via amniocentesis of chorionic villus sampling.
MRI
In some children without "classic" holoprosencephaly, microforms of holoprosencephaly may be noted on MRI, including missing olfactory tracts and bulbs and absent or hypoplastic corpus callosum.
Treatment
At present, treatment for 18p- is symptomatic, meaning that the focus is on treating the signs and symptoms of the conditions as they arise. To ensure early diagnosis and treatment, it is suggested that people with 18p- undergo routine screenings for hearing and vision problems.
Terminology
The preferred terminology for this condition is 18p-. In the past, it has been referred to as partial monosomy 18p and, rarely, as "de Grouchy syndrome, type 1".
Research
Currently, research is focusing on identifying the role of the genes on 18p in causing the signs and symptoms associated with deletions of 18p. This will ultimately enable predictive genotyping.
TGIF-Mutations and deletions of this gene have been associated with holoprosencephaly. Penetrance is incomplete, meaning that a deletion of one copy of this gene is not in and of itself sufficient to cause holoprosencephaly. Ten to fifteen percent of people with 18p- have holoprosencephaly, suggesting that other genetic and environmental facts play a role in the etiology of holoprosencephaly in these individuals.
References
- Turleau, Catherine (2008). "Monosomy 18p". Orphanet Journal of Rare Diseases. 3 (1): 4. doi:10.1186/1750-1172-3-4. ISSN 1750-1172. PMC 2265258. PMID 18284672.
- Hasi-Zogaj M, Sebold C, Heard P, Carter E, Soileau B, Hill A, Rupert D, Perry B, Atkinson S, O'Donnell L, Gelfond J, Lancaster J, Fox PT, Hale DE, Cody JD (2015). "A review of 18p deletions". Am J Med Genet C Semin Med Genet. 169 (3): 251–64. doi:10.1002/ajmg.c.31445. PMID 26250845. S2CID 20556694.