Oncogene
An oncogene is a gene that has the potential to cause cancer.[1] In tumor cells, these genes are often mutated, or expressed at high levels.[2]
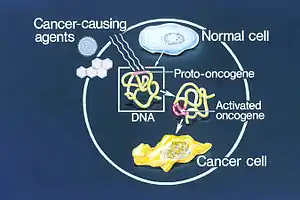
Most normal cells will undergo a programmed form of rapid cell death (apoptosis) when critical functions are altered and malfunctioning. Activated oncogenes can cause those cells designated for apoptosis to survive and proliferate instead.[3] Most oncogenes began as proto-oncogenes: normal genes involved in cell growth and proliferation or inhibition of apoptosis. If, through mutation, normal genes promoting cellular growth are up-regulated (gain-of-function mutation), they will predispose the cell to cancer; thus, they are termed "oncogenes". Usually multiple oncogenes, along with mutated apoptotic or tumor suppressor genes will all act in concert to cause cancer. Since the 1970s, dozens of oncogenes have been identified in human cancer. Many cancer drugs target the proteins encoded by oncogenes.[2][4][5][6]
History
The theory of oncogenes was foreshadowed by the German biologist Theodor Boveri in his 1914 book Zur Frage der Entstehung Maligner Tumoren (Concerning the Origin of Malignant Tumors) in which he predicted the existence of oncogenes (Teilungsfoerdernde Chromosomen) that become amplified (im permanenten Übergewicht) during tumor development.[7]
Later on, the term "oncogene" was rediscovered in 1969 by National Cancer Institute scientists George Todaro and Robert Huebner.[8]
The first confirmed oncogene was discovered in 1970 and was termed SRC (pronounced "sarc" as it is short for sarcoma). SRC was first discovered as an oncogene in a chicken retrovirus. Experiments performed by Dr. G. Steve Martin of the University of California, Berkeley demonstrated that SRC was indeed the gene of the virus that acted as an oncogene upon infection.[9] The first nucleotide sequence of v-Src was sequenced in 1980 by A.P. Czernilofsky et al.[10]
In 1976, Drs. Dominique Stéhelin, J. Michael Bishop and Harold E. Varmus of the University of California, San Francisco demonstrated that oncogenes were activated proto-oncogenes as is found in many organisms, including humans. Bishop and Varmus were awarded the Nobel Prize in Physiology or Medicine in 1989 for their discovery of the cellular origin of retroviral oncogenes.[11]
Dr. Robert Weinberg is credited with discovering the first identified human oncogene in a human bladder cancer cell line.[12][13] The molecular nature of the mutation leading to oncogenesis was subsequently isolated and characterized by the Spanish biochemist Mariano Barbacid and published in Nature in 1982.[14] Dr. Barbacid spent the following months extending his research, eventually discovering that the oncogene was a mutated allele of HRAS and characterizing its activation mechanism.
The resultant protein encoded by an oncogene is termed oncoprotein.[15] Oncogenes play an important role in the regulation or synthesis of proteins linked to tumorigenic cell growth. Some oncoproteins are accepted and used as tumor markers.
Proto-oncogene
A proto-oncogene is a normal gene that could become an oncogene due to mutations or increased expression. Proto-oncogenes code for proteins that help to regulate the cell growth and differentiation. Proto-oncogenes are often involved in signal transduction and execution of mitogenic signals, usually through their protein products. Upon acquiring an activating mutation, a proto-oncogene becomes a tumor-inducing agent, an oncogene.[16] Examples of proto-oncogenes include RAS, WNT, MYC, ERK, and TRK. The MYC gene is implicated in Burkitt's lymphoma, which starts when a chromosomal translocation moves an enhancer sequence within the vicinity of the MYC gene. The MYC gene codes for widely used transcription factors. When the enhancer sequence is wrongly placed, these transcription factors are produced at much higher rates. Another example of an oncogene is the Bcr-Abl gene found on the Philadelphia chromosome, a piece of genetic material seen in Chronic Myelogenous Leukemia caused by the translocation of pieces from chromosomes 9 and 22. Bcr-Abl codes for a tyrosine kinase, which is constitutively active, leading to uncontrolled cell proliferation. (More information about the Philadelphia Chromosome below)
Activation

The proto-oncogene can become an oncogene by a relatively small modification of its original function. There are three basic methods of activation:
- A mutation within a proto-oncogene, or within a regulatory region (for example the promoter region), can cause a change in the protein structure, causing
- an increase in protein (enzyme) activity
- a loss of regulation
- An increase in the amount of a certain protein (protein concentration), caused by
- an increase of protein expression (through misregulation)
- an increase of protein (mRNA) stability, prolonging its existence and thus its activity in the cell
- gene duplication (one type of chromosome abnormality), resulting in an increased amount of protein in the cell
- A chromosomal translocation (another type of chromosome abnormality)
- There are 2 different types of chromosomal translocations that can occur:
- translocation events which relocate a proto-oncogene to a new chromosomal site that leads to higher expression
- translocation events that lead to a fusion between a proto-oncogene and a 2nd gene (this creates a fusion protein with increased cancerous/oncogenic activity)
- the expression of a constitutively active hybrid protein. This type of mutation in a dividing stem cell in the bone marrow leads to adult leukemia
- Philadelphia Chromosome is an example of this type of translocation event. This chromosome was discovered in 1960 by Peter Nowell and David Hungerford, and it is a fusion of parts of DNA from chromosome 22 and chromosome 9. The broken end of chromosome 22 contains the "BCR" gene, which fuses with a fragment of chromosome 9 that contains the "ABL1" gene. When these two chromosome fragments fuse the genes also fuse creating a new gene: "BCR-ABL". This fused gene encodes for a protein that displays high protein tyrosine kinase activity (this activity is due to the "ABL1" half of the protein). The unregulated expression of this protein activates other proteins that are involved in cell cycle and cell division which can cause a cell to grow and divide uncontrollably (the cell becomes cancerous). As a result, the Philadelphia Chromosome is associated with Chronic Myelogenous Leukemia (as mentioned before) as well as other forms of Leukemia.[17]
The expression of oncogenes can be regulated by microRNAs (miRNAs), small RNAs 21-25 nucleotides in length that control gene expression by downregulating them.[18] Mutations in such microRNAs (known as oncomirs) can lead to activation of oncogenes.[19] Antisense messenger RNAs could theoretically be used to block the effects of oncogenes.
Classification
There are several systems for classifying oncogenes,[20] but there is not yet a widely accepted standard. They are sometimes grouped both spatially (moving from outside the cell inwards) and chronologically (parallelling the "normal" process of signal transduction). There are several categories that are commonly used:
| Category | Examples | Cancers | Gene functions |
| Growth factors, or mitogens | c-Sis | glioblastomas, fibrosarcomas, osteosarcomas, breast carcinomas, and melanomas[21] | induces cell proliferation. |
| Receptor tyrosine kinases | epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), and vascular endothelial growth factor receptor (VEGFR), HER2/neu | Breast cancer, gastrointestinal stromal tumours, non-small-cell lung cancer and pancreatic cancer[22] | transduce signals for cell growth and differentiation. |
| Cytoplasmic tyrosine kinases | Src-family, Syk-ZAP-70 family, and BTK family of tyrosine kinases, the Abl gene in CML - Philadelphia chromosome | colorectal and breast cancers, melanomas, ovarian cancers, gastric cancers, head and neck cancers, pancreatic cancer, lung cancer, brain cancers, and blood cancers[23] | mediate the responses to, and the activation receptors of cell proliferation, migration, differentiation, and survival[24] |
| Cytoplasmic Serine/threonine kinases and their regulatory subunits | Raf kinase, and cyclin-dependent kinases (through overexpression). | malignant melanoma, papillary thyroid cancer, colorectal cancer, and ovarian cancer[25] | involved in organism development, cell cycle regulation, cell proliferation, differentiation, cells survival, and apoptosis[26] |
| Regulatory GTPases | Ras protein | adenocarcinomas of the pancreas and colon, thyroid tumors, and myeloid leukemia[27] | involved in signalling a major pathway leading to cell proliferation.[28] |
| Transcription factors | myc gene | malignant T-cell lymphomas and acute myeloid leukemias, breast cancer, pancreatic cancer, retinoblastoma, and small cell lung cancer[29] | regulate transcription of genes that induce cell proliferation. |
Additional oncogenetic regulator properties include:
- Growth factors are usually secreted by either specialized or non-specialized cells to induce cell proliferation in themselves, nearby cells, or distant cells. An oncogene may cause a cell to secrete growth factors even though it does not normally do so. It will thereby induce its own uncontrolled proliferation (autocrine loop), and proliferation of neighboring cells, possibly leading to tumor formation. It may also cause production of growth hormones in other parts of the body.
- Receptor tyrosine kinases add phosphate groups to other proteins in order to turn them on or off. Receptor kinases add phosphate groups to receptor proteins at the surface of the cell (which receives protein signals from outside the cell and transmits them to the inside of the cell). Tyrosine kinases add phosphate groups to the amino acid tyrosine in the target protein. They can cause cancer by turning the receptor permanently on (constitutively), even without signals from outside the cell.
- Ras is a small GTPase that hydrolyses GTP into GDP and phosphate. Ras is activated by growth factor signaling (i.e., EGF, TGFbeta) and acting as a binary switch (on/off) in growth signaling pathways. Downstream effectors of Ras include three mitogen-activated protein kinases Raf a MAP Kinase Kinase Kinase (MAPKKK), MEK a MAP Kinase Kinase (MAPKK), and ERK a MAP Kinase(MAPK), which in turn regulate genes that mediate cell proliferation.[30]
See also
- Anticancer gene
- Oncogenomics
- Tumor suppressor gene
- Oncovirus
- Genetic predisposition
- Quantitative trait locus
- Genetic susceptibility
- Oncometabolism
References
- Wilbur B, ed. (2009). The World of the Cell (7th ed.). San Francisco, California.
- Kimball's Biology Pages. "Oncogenes" Free full text
- The Nobel Prize in Physiology or Medicine 2002. Illustrated presentation.
- Croce CM (January 2008). "Oncogenes and cancer". The New England Journal of Medicine. 358 (5): 502–511. doi:10.1056/NEJMra072367. PMID 18234754.
- Yokota J (March 2000). "Tumor progression and metastasis". Carcinogenesis. 21 (3): 497–503. doi:10.1093/carcin/21.3.497. PMID 10688870.
- The Nobel Prize in Physiology or Medicine 1989 to J. Michael Bishop and Harold E. Varmus for their discovery of "the cellular origin of retroviral oncogenes".
- Boveri T (1914). Zur Frage der Entstehung maligner Tumoren. Jena: Gustav Fischer.
- The Emperor of All Maladies, Siddhartha Mukherjee, 2011, p. 363
- Martin GS (June 2001). "The hunting of the Src". Nature Reviews. Molecular Cell Biology. 2 (6): 467–475. doi:10.1038/35073094. PMID 11389470. S2CID 205016442.
- Czernilofsky AP, Levinson AD, Varmus HE, Bishop JM, Tischer E, Goodman HM (September 1980). "Nucleotide sequence of an avian sarcoma virus oncogene (src) and proposed amino acid sequence for gene product". Nature. 287 (5779): 198–203. Bibcode:1980Natur.287..198C. doi:10.1038/287198a0. PMID 6253794. S2CID 4231060.
- Nobel Prize in Physiology or Medicine for 1989 jointly to J. Michael Bishop and Harold E. Varmus for their discovery of "the cellular origin of retroviral oncogenes". Press Release.
- Shih C, Weinberg RA (May 1982). "Isolation of a transforming sequence from a human bladder carcinoma cell line". Cell. 29 (1): 161–169. doi:10.1016/0092-8674(82)90100-3. PMID 6286138. S2CID 12046552.
- Lowry F (5 May 2011). "Robert Weinberg Rewarded for Oncogene Discovery". Medscape. Retrieved 6 February 2020.
- Reddy EP, Reynolds RK, Santos E, Barbacid M (November 1982). "A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene". Nature. 300 (5888): 149–152. Bibcode:1982Natur.300..149R. doi:10.1038/300149a0. PMID 7133135. S2CID 34599264.
- Mitchell RS, Kumar V, Abbas AK, Fausto N (2007). "Chapter 20 - Neoplasms of the Thyroid". Robbins Basic Pathology (8th ed.). Philadelphia: Saunders. ISBN 978-1-4160-2973-1.
- Todd R, Wong DT (1999). "Oncogenes". Anticancer Research. 19 (6A): 4729–4746. PMID 10697588.
- Chial H (2008). "Proto-oncogenes to Oncogenes to Cancer". Nature Education. 1 (1).
- Negrini M, Ferracin M, Sabbioni S, Croce CM (June 2007). "MicroRNAs in human cancer: from research to therapy". Journal of Cell Science. 120 (Pt 11): 1833–1840. doi:10.1242/jcs.03450. PMID 17515481.
- Esquela-Kerscher A, Slack FJ (April 2006). "Oncomirs - microRNAs with a role in cancer". Nature Reviews. Cancer. 6 (4): 259–269. doi:10.1038/nrc1840. PMID 16557279. S2CID 10620165.
- THE Medical Biochemistry Page
- Press RD, Misra A, Gillaspy G, Samols D, Goldthwait DA (June 1989). "Control of the expression of c-sis mRNA in human glioblastoma cells by phorbol ester and transforming growth factor beta 1". Cancer Research. 49 (11): 2914–2920. PMID 2655888.
- Gschwind A, Fischer OM, Ullrich A (May 2004). "The discovery of receptor tyrosine kinases: targets for cancer therapy". Nature Reviews. Cancer. 4 (5): 361–370. doi:10.1038/nrc1360. PMID 15122207. S2CID 6939454.
- Summy JM, Gallick GE (December 2003). "Src family kinases in tumor progression and metastasis". Cancer and Metastasis Reviews. 22 (4): 337–358. doi:10.1023/A:1023772912750. PMID 12884910. S2CID 12380282.
- Thomas SM, Brugge JS (1 November 1997). "Cellular functions regulated by Src family kinases". Annual Review of Cell and Developmental Biology. 13 (1): 513–609. doi:10.1146/annurev.cellbio.13.1.513. PMID 9442882.
- Garnett MJ, Marais R (October 2004). "Guilty as charged: B-RAF is a human oncogene". Cancer Cell. 6 (4): 313–319. doi:10.1016/j.ccr.2004.09.022. PMID 15488754.
- Leicht DT, Balan V, Kaplun A, Singh-Gupta V, Kaplun L, Dobson M, Tzivion G (August 2007). "Raf kinases: function, regulation and role in human cancer". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1773 (8): 1196–1212. doi:10.1016/j.bbamcr.2007.05.001. PMC 1986673. PMID 17555829.
- Bos JL (September 1989). "ras oncogenes in human cancer: a review". Cancer Research. 49 (17): 4682–4689. PMID 2547513.
- Hilgenfeld R (December 1995). "Regulatory GTPases". Current Opinion in Structural Biology. 5 (6): 810–817. doi:10.1016/0959-440X(95)80015-8. PMID 8749370.
- Felsher DW, Bishop JM (August 1999). "Reversible tumorigenesis by MYC in hematopoietic lineages". Molecular Cell. 4 (2): 199–207. doi:10.1016/S1097-2765(00)80367-6. PMID 10488335.
- Cargnello M, Roux PP (March 2011). "Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases". Microbiology and Molecular Biology Reviews. 75 (1): 50–83. doi:10.1128/MMBR.00031-10. PMC 3063353. PMID 21372320.