Computational biology
Computational biology refers to the use of data analysis, mathematical modeling and computational simulations to understand biological systems and relationships.[1] An intersection of computer science, biology, and big data, the field also has foundations in applied mathematics, chemistry, and genetics.[2] It differs from biological computing, a subfield of computer engineering which uses bioengineering to build computers.
.jpg.webp)
History
Bioinformatics, the analysis of informatics processes in biological systems, began in the early 1970s. At this time, research in artificial intelligence was using network models of the human brain in order to generate new algorithms. This use of biological data pushed biological researchers to use computers to evaluate and compare large data sets in their own field.[3]
By 1982, researchers shared information via punch cards. The amount of data grew exponentially by the end of the 1980s, requiring new computational methods for quickly interpreting relevant information.[3]
Perhaps the best-known example of computational biology, the Human Genome Project, officially began in 1990.[4] By 2003, the project had mapped around 85% of the human genome, satisfying its initial goals.[5] Work continued, however, and by 2021 level "complete genome" was reached with only 0.3% remaining bases covered by potential issues.[6][7] The missing Y chromosome was added in January 2022.
Since the late 1990s, computational biology has become an important part of biology, leading to numerous subfields.[8] Today, the International Society for Computational Biology recognizes 21 different 'Communities of Special Interest', each representing a slice of the larger field.[9] In addition to helping sequence the human genome, computational biology has helped create accurate models of the human brain, map the 3D structure of genomes, and model biological systems.[3]
Applications
Anatomy
Computational anatomy is the study of anatomical shape and form at the visible or gross anatomical scale of morphology. It involves the development of computational mathematical and data-analytical methods for modeling and simulating biological structures. It focuses on the anatomical structures being imaged, rather than the medical imaging devices. Due to the availability of dense 3D measurements via technologies such as magnetic resonance imaging, computational anatomy has emerged as a subfield of medical imaging and bioengineering for extracting anatomical coordinate systems at the morpheme scale in 3D.

The original formulation of computational anatomy is as a generative model of shape and form from exemplars acted upon via transformations.[10] The diffeomorphism group is used to study different coordinate systems via coordinate transformations as generated via the Lagrangian and Eulerian velocities of flow from one anatomical configuration in to another. It relates with shape statistics and morphometrics, with the distinction that diffeomorphisms are used to map coordinate systems, whose study is known as diffeomorphometry.

Data and modeling
Mathematical biology is the use of mathematical models of living organisms to examine the systems that govern structure, development, and behavior in biological systems. This entails a more theoretical approach to problems, rather than its more empirically-minded counterpart of experimental biology.[11] Mathematical biology draws on discrete mathematics, topology (also useful for computational modeling), Bayesian statistics, linear algebra and Boolean algebra.[12]
These mathematical approaches have enabled the creation of databases and other methods for storing, retrieving, and analyzing biological data, a field known as bioinformatics. Usually, this process involves genetics and analyzing genes.
Gathering and analyzing large datasets have made way for growing research fields such as data mining,[12] and computational biomodeling, which refers to building computer models and visual simulations of biological systems. This allows researchers to predict how such systems will react to different environments, useful for determining if a system can "maintain their state and functions against external and internal perturbations".[13] While current techniques focus on small biological systems, researchers are working on approaches that will allow for larger networks to be analyzed and modeled. A majority of researchers believe that this will be essential in developing modern medical approaches to creating new drugs and gene therapy.[13] A useful modeling approach is to use Petri nets via tools such as esyN.[14]
Along similar lines, until recent decades theoretical ecology has largely dealt with analytic models that were detached from the statistical models used by empirical ecologists. However, computational methods have aided in developing ecological theory via simulation of ecological systems, in addition to increasing application of methods from computational statistics in ecological analyses.
Systems Biology
Systems biology consists of computing the interactions between various biological systems ranging from the cellular level to entire populations with the goal of discovering emergent properties. This process usually involves networking cell signaling and metabolic pathways. Systems biology often uses computational techniques from biological modeling and graph theory to study these complex interactions at cellular levels.[12]
Evolutionary biology
Computational biology has assisted evolutionary biology by:
- Using DNA data to reconstruct the tree of life with computational phylogenetics
- Fitting population genetics models (either forward time[15] or backward time) to DNA data to make inferences about demographic or selective history
- Building population genetics models of evolutionary systems from first principles in order to predict what is likely to evolve
Genomics

Computational genomics is the study of the genomes of cells and organisms. The Human Genome Project is one example of computational genomics. This project looks to sequence the entire human genome into a set of data. Once fully implemented, this could allow for doctors to analyze the genome of an individual patient.[16] This opens the possibility of personalized medicine, prescribing treatments based on an individual's pre-existing genetic patterns. Researchers are looking to sequence the genomes of animals, plants, bacteria, and all other types of life.[17]
One of the main ways that genomes are compared is by sequence homology. Homology is the study of biological structures and nucleotide sequences in different organisms that come from a common ancestor. Research suggests that between 80 and 90% of genes in newly sequenced prokaryotic genomes can be identified this way.[17]
Sequence alignment is another process for comparing and detecting similarities between biological sequences or genes. Sequence alignment is useful in a number of bioinformatics applications, such as computing the longest common subsequence of two genes or comparing variants of certain diseases.[18]
An untouched project in computational genomics is the analysis of intergenic regions, which comprise roughly 97% of the human genome.[17] Researchers are working to understand the functions of non-coding regions of the human genome through the development of computational and statistical methods and via large consortia projects such as ENCODE and the Roadmap Epigenomics Project.
Understanding how individual genes contribute to the biology of an organism at the molecular, cellular, and organism levels is known as gene ontology. The Gene Ontology Consortium's mission is to develop an up-to-date, comprehensive, computational model of biological systems, from the molecular level to larger pathways, cellular, and organism-level systems. The Gene Ontology resource provides a computational representation of current scientific knowledge about the functions of genes (or, more properly, the protein and non-coding RNA molecules produced by genes) from many different organisms, from humans to bacteria.[19]
3D genomics is a subsection in computational biology that focuses on the organization and interaction of genes within a eukaryotic cell. One method used to gather 3D genomic data is through Genome Architecture Mapping (GAM). GAM measures 3D distances of chromatin and DNA in the genome by combining cryosectioning, the process of cutting a strip from the nucleus to examine the DNA, with laser microdissection. A nuclear profile is simply this strip or slice that is taken from the nucleus. Each nuclear profile contains genomic windows, which are certain sequences of nucleotides - the base unit of DNA. GAM captures a genome network of complex, multi enhancer chromatin contacts throughout a cell.[20]
Neuroscience
Computational neuroscience is the study of brain function in terms of the information processing properties of the nervous system. A subset of neuroscience, it looks to model the brain to examine specific aspects of the neurological system.[21] Models of the brain include:
- Realistic Brain Models: These models look to represent every aspect of the brain, including as much detail at the cellular level as possible. Realistic models provide the most information about the brain, but also have the largest margin for error. More variables in a brain model create the possibility for more error to occur. These models do not account for parts of the cellular structure that scientists do not know about. Realistic brain models are the most computationally heavy and the most expensive to implement.[22]
- Simplifying Brain Models: These models look to limit the scope of a model in order to assess a specific physical property of the neurological system. This allows for the intensive computational problems to be solved, and reduces the amount of potential error from a realistic brain model.[22]
It is the work of computational neuroscientists to improve the algorithms and data structures currently used to increase the speed of such calculations.
Computational neuropsychiatry is an emerging field that uses mathematical and computer-assisted modeling of brain mechanisms involved in mental disorders. Several initiatives have demonstrated that computational modeling is an important contribution to understand neuronal circuits that could generate mental functions and dysfunctions.[23][24][25]
Pharmacology
Computational pharmacology is "the study of the effects of genomic data to find links between specific genotypes and diseases and then screening drug data".[26] The pharmaceutical industry requires a shift in methods to analyze drug data. Pharmacologists were able to use Microsoft Excel to compare chemical and genomic data related to the effectiveness of drugs. However, the industry has reached what is referred to as the Excel barricade. This arises from the limited number of cells accessible on a spreadsheet. This development led to the need for computational pharmacology. Scientists and researchers develop computational methods to analyze these massive data sets. This allows for an efficient comparison between the notable data points and allows for more accurate drugs to be developed.[27]
Analysts project that if major medications fail due to patents, that computational biology will be necessary to replace current drugs on the market. Doctoral students in computational biology are being encouraged to pursue careers in industry rather than take Post-Doctoral positions. This is a direct result of major pharmaceutical companies needing more qualified analysts of the large data sets required for producing new drugs.[27]
Similarly, computational oncology aims to determine the future mutations in cancer through algorithmic approaches. Research in this field has led to the use of high-throughput measurement that millions of data points using robotics and other sensing devices. This data is collected from DNA, RNA, and other biological structures. Areas of focus include determining the characteristics of tumors, analyzing molecules that are deterministic in causing cancer, and understanding how the human genome relates to the causation of tumors and cancer.[28][29]
Techniques
Computational biologists use a wide range of software and algorithms to carry out their research.
Unsupervised Learning
Unsupervised learning is a type of algorithm that finds patterns in unlabeled data. One example is k-means clustering, which aims to partition n data points into k clusters, in which each data point belongs to the cluster with the nearest mean. Another version is the k-medoids algorithm, which, when selecting a cluster center or cluster centroid, will pick one of its data points in the set, and not just an average of the cluster.
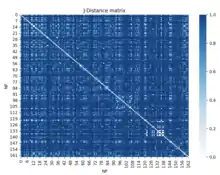
The algorithm follows these steps:
- Randomly select k distinct data points. These are the initial clusters.
- Measure the distance between each point and each of the 'k' clusters. (This is the distance of the points from each point k).
- Assign each point to the nearest cluster.
- Find the center of each cluster (medoid).
- Repeat until the clusters no longer change.
- Assess the quality of the clustering by adding up the variation within each cluster.
- Repeat the processes with different values of k.
- Pick the best value for 'k' by finding the "elbow" in the plot of which k value has the lowest variance.
One example of this in biology is used in the 3D mapping of a genome. Information of a mouse's HIST1 region of chromosome 13 is gathered from Gene Expression Omnibus.[30] This information contains data on which nuclear profiles show up in certain genomic regions. With this information, the Jaccard distance can be used to find a normalized distance between all the loci.
Graph Analytics
Graph analytics, or network analysis, is the study of graphs that represent connections between different objects. Graphs can represent all kinds of networks in biology such as Protein-protein interaction networks, regulatory networks, Metabolic and biochemical networks and much more. There are many ways to analyze these networks. One of which is looking at Centrality in graphs. Finding centrality in graphs assigns nodes rankings to their popularity or centrality in the graph. This can be useful in finding what nodes are most important. This can be very useful in biology in many ways. For example, if we were to have data on the activity of genes in a given time period, we can use degree centrality to see what genes are most active throughout the network, or what genes interact with others the most throughout the network. This can help us understand what roles certain genes play in the network.
There are many ways to calculate centrality in graphs all of which can give different kinds of information on centrality. Finding centralities in biology can be applied in many different circumstances, some of which are gene regulatory, protein interaction and metabolic networks.[31]
Supervised Learning
Supervised learning is a type of algorithm that learns from labeled data and learns how to assign labels to future data that is unlabeled. In biology supervised learning can be helpful when we have data that we know how to categorize and we would like to categorize more data into those categories.
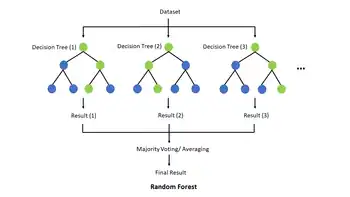
A common supervised learning algorithm is the random forest, which uses numerous decision trees to train a model to classify a dataset. Forming the basis of the random forest, a decision tree is a structure which aims to classify, or label, some set of data using certain known features of that data. A practical biological example of this would be taking an individual's genetic data and predicting whether or not that individual is predisposed to develop a certain disease or cancer. At each internal node the algorithm checks the dataset for exactly one feature, a specific gene in the previous example, and then branches left or right based on the result. Then at each leaf node, the decision tree assigns a class label to the dataset. So in practice, the algorithm walks a specific root-to-leaf path based on the input dataset through the decision tree, which results in the classification of that dataset. Commonly, decision trees have target variables that take on discrete values, like yes/no, in which case it is referred to as a classification tree, but if the target variable is continuous then it is called a regression tree. To construct a decision tree, it must first be trained using a training set to identify which features are the best predictors of the target variable.
Open source software
Open source software provides a platform for computational biology where everyone can access and benefit from software developed in research. PLOS cites four main reasons for the use of open source software:
- Reproducibility: This allows for researchers to use the exact methods used to calculate the relations between biological data.
- Faster development: developers and researchers do not have to reinvent existing code for minor tasks. Instead they can use pre-existing programs to save time on the development and implementation of larger projects.
- Increased quality: Having input from multiple researchers studying the same topic provides a layer of assurance that errors will not be in the code.
- Long-term availability: Open source programs are not tied to any businesses or patents. This allows for them to be posted to multiple web pages and ensure that they are available in the future.[32]
Research
There are several large conferences that are concerned with computational biology. Some notable examples are Intelligent Systems for Molecular Biology, European Conference on Computational Biology and Research in Computational Molecular Biology.
There are also numerous journals dedicated to computational biology. Some notable examples include Journal of Computational Biology and PLOS Computational Biology, a peer-reviewed open access journal that has many notable research projects in the field of computational biology. They provide reviews on software, tutorials for open source software, and display information on upcoming computational biology conferences.
Related fields
Computational biology, bioinformatics and mathematical biology are all interdisciplinary approaches to the life sciences that draw from quantitative disciplines such as mathematics and information science. The NIH describes computational/mathematical biology as the use of computational/mathematical approaches to address theoretical and experimental questions in biology and, by contrast, bioinformatics as the application of information science to understand complex life-sciences data.[1]
Specifically, the NIH defines
Computational biology: The development and application of data-analytical and theoretical methods, mathematical modeling and computational simulation techniques to the study of biological, behavioral, and social systems.[1]
Bioinformatics: Research, development, or application of computational tools and approaches for expanding the use of biological, medical, behavioral or health data, including those to acquire, store, organize, archive, analyze, or visualize such data.[1]
While each field is distinct, there may be significant overlap at their interface,[1] so much so that to many, bioinformatics and computational biology are terms that are used interchangeably.
The terms computational biology and evolutionary computation have a similar name, but are not to be confused. Unlike computational biology, evolutionary computation is not concerned with modeling and analyzing biological data. It instead creates algorithms based on the ideas of evolution across species. Sometimes referred to as genetic algorithms, the research of this field can be applied to computational biology. While evolutionary computation is not inherently a part of computational biology, computational evolutionary biology is a subfield of it.[33]
See also
- Artificial life
- Bioinformatics
- Biological computing
- Biological simulation
- Biosimulation
- Biostatistics
- Computational audiology
- Computational chemistry
- Computational science
- Computational history
- DNA sequencing
- Functional genomics
- International Society for Computational Biology
- List of bioinformatics institutions
- List of biological databases
- Mathematical biology
- Monte Carlo method
- Molecular modeling
- Network biology
- Phylogenetics
- Proteomics
- Structural genomics
- Synthetic biology
- Systems biology
References
- "NIH working definition of bioinformatics and computational biology" (PDF). Biomedical Information Science and Technology Initiative. 17 July 2000. Archived from the original (PDF) on 5 September 2012. Retrieved 18 August 2012.
- "About the CCMB". Center for Computational Molecular Biology. Retrieved 18 August 2012.
- Hogeweg, Paulien (7 March 2011). "The Roots of Bioinformatics in Theoretical Biology". PLOS Computational Biology. 3. 7 (3): e1002021. Bibcode:2011PLSCB...7E2021H. doi:10.1371/journal.pcbi.1002021. PMC 3068925. PMID 21483479.
- "The Human Genome Project". The Human Genome Project. 22 December 2020. Retrieved 13 April 2022.
- "Human Genome Project FAQ". Genome.gov. Retrieved 2022-04-20.
- "T2T-CHM13v1.1 - Genome - Assembly - NCBI". www.ncbi.nlm.nih.gov. Retrieved 2022-04-20.
- "Genome List - Genome - NCBI". www.ncbi.nlm.nih.gov. Retrieved 2022-04-20.
- Bourne, Philip (2012). "Rise and Demise of Bioinformatics? Promise and Progress". PLOS Computational Biology. 8 (4): e1002487. Bibcode:2012PLSCB...8E2487O. doi:10.1371/journal.pcbi.1002487. PMC 3343106. PMID 22570600.
- "COSI Information". www.iscb.org. Retrieved 2022-04-21.
- Grenander, Ulf; Miller, Michael I. (1998-12-01). "Computational Anatomy: An Emerging Discipline". Q. Appl. Math. 56 (4): 617–694. doi:10.1090/qam/1668732.
- "Mathematical Biology | Faculty of Science". www.ualberta.ca. Retrieved 2022-04-18.
- "The Sub-fields of Computational Biology". Ninh Laboratory of Computational Biology. 2013-02-18. Retrieved 2022-04-18.
- Kitano, Hiroaki (14 November 2002). "Computational systems biology". Nature. 420 (6912): 206–10. Bibcode:2002Natur.420..206K. doi:10.1038/nature01254. PMID 12432404. S2CID 4401115. ProQuest 204483859.
- Favrin, Bean (2 September 2014). "esyN: Network Building, Sharing and Publishing". PLOS ONE. 9 (9): e106035. Bibcode:2014PLoSO...9j6035B. doi:10.1371/journal.pone.0106035. PMC 4152123. PMID 25181461.
- Antonio Carvajal-Rodríguez (2012). "Simulation of Genes and Genomes Forward in Time". Current Genomics. 11 (1): 58–61. doi:10.2174/138920210790218007. PMC 2851118. PMID 20808525.
- "Genome Sequencing to the Rest of Us". Scientific American.
- Koonin, Eugene (6 March 2001). "Computational Genomics". Curr. Biol. 11 (5): 155–158. doi:10.1016/S0960-9822(01)00081-1. PMID 11267880. S2CID 17202180.
- "Sequence Alignment - an overview | ScienceDirect Topics". www.sciencedirect.com. Retrieved 2022-04-18.
- "Gene Ontology Resource". Gene Ontology Resource. Retrieved 2022-04-18.
- Beagrie, Robert A.; Scialdone, Antonio; Schueler, Markus; Kraemer, Dorothee C. A.; Chotalia, Mita; Xie, Sheila Q.; Barbieri, Mariano; de Santiago, Inês; Lavitas, Liron-Mark; Branco, Miguel R.; Fraser, James (March 2017). "Complex multi-enhancer contacts captured by genome architecture mapping". Nature. 543 (7646): 519–524. Bibcode:2017Natur.543..519B. doi:10.1038/nature21411. ISSN 1476-4687. PMC 5366070. PMID 28273065.
- "Computational Neuroscience | Neuroscience". www.bu.edu.
- Sejnowski, Terrence; Christof Koch; Patricia S. Churchland (9 September 1988). "Computational Neuroscience". Science. 4871. 241 (4871): 1299–306. Bibcode:1988Sci...241.1299S. doi:10.1126/science.3045969. PMID 3045969.
- Dauvermann, Maria R.; Whalley, Heather C.; Schmidt, Andrã©; Lee, Graham L.; Romaniuk, Liana; Roberts, Neil; Johnstone, Eve C.; Lawrie, Stephen M.; Moorhead, Thomas W. J. (2014). "Computational Neuropsychiatry – Schizophrenia as a Cognitive Brain Network Disorder". Frontiers in Psychiatry. 5: 30. doi:10.3389/fpsyt.2014.00030. PMC 3971172. PMID 24723894.
- Tretter, F.; Albus, M. (December 2007). "'Computational Neuropsychiatry' of Working Memory Disorders in Schizophrenia: The Network Connectivity in Prefrontal Cortex - Data and Models". Pharmacopsychiatry. 40 (S 1): S2–S16. doi:10.1055/S-2007-993139. S2CID 18574327.
- Marin-Sanguino, A.; Mendoza, E. (2008). "Hybrid Modeling in Computational Neuropsychiatry". Pharmacopsychiatry. 41: S85–S88. doi:10.1055/s-2008-1081464. PMID 18756425.
- Price, Michael (2012-04-13). "Computational Biologists: The Next Pharma Scientists?".
- Jessen, Walter (2012-04-15). "Pharma's shifting strategy means more jobs for computational biologists".
- Barbolosi, Dominique; Ciccolini, Joseph; Lacarelle, Bruno; Barlesi, Fabrice; Andre, Nicolas (2016). "Computational oncology--mathematical modelling of drug regimens for precision medicine". Nature Reviews Clinical Oncology. 13 (4): 242–254. doi:10.1038/nrclinonc.2015.204. PMID 26598946. S2CID 22492353.
- Yakhini, Zohar (2011). "Cancer Computational Biology". BMC Bioinformatics. 12: 120. doi:10.1186/1471-2105-12-120. PMC 3111371. PMID 21521513.
- link
- Koschützki, Dirk; Schreiber, Falk (2008-05-15). "Centrality Analysis Methods for Biological Networks and Their Application to Gene Regulatory Networks". Gene Regulation and Systems Biology. 2: 193–201. doi:10.4137/grsb.s702. ISSN 1177-6250. PMC 2733090. PMID 19787083.
- Prlić, Andreas; Lapp, Hilmar (2012). "The PLOS Computational Biology Software Section". PLOS Computational Biology. 8 (11): e1002799. Bibcode:2012PLSCB...8E2799P. doi:10.1371/journal.pcbi.1002799. PMC 3510099.
- Foster, James (June 2001). "Evolutionary Computation". Nature Reviews Genetics. 2 (6): 428–436. doi:10.1038/35076523. PMID 11389459. S2CID 205017006.
External links

| Databases |
|
|---|---|
| Software |
|
| Other |
|
| Institutions |
|
| Organizations |
|
| Meetings |
|
| File formats |
|
| Related topics |
|
| |