Creosote
Creosote is a category of carbonaceous chemicals formed by the distillation of various tars and pyrolysis of plant-derived material, such as wood or fossil fuel. They are typically used as preservatives or antiseptics.[2]

Some creosote types were used historically as a treatment for components of seagoing and outdoor wood structures to prevent rot (e.g., bridgework and railroad ties, see image). Samples may be found commonly inside chimney flues, where the coal or wood burns under variable conditions, producing soot and tarry smoke. Creosotes are the principal chemicals responsible for the stability, scent, and flavor characteristic of smoked meat; the name is derived from Greek κρέας (kreas) 'meat', and σωτήρ (sōtēr) 'preserver'.[3]
The two main kinds recognized in industry are coal-tar creosote and wood-tar creosote. The coal-tar variety, having stronger and more toxic properties, has chiefly been used as a preservative for wood; coal-tar creosote was also formerly used as an escharotic, to burn malignant skin tissue, and in dentistry, to prevent necrosis, before its carcinogenic properties became known.[4][5] The wood-tar variety has been used for meat preservation, ship treatment, and such medical purposes as an anaesthetic, antiseptic, astringent, expectorant, and laxative, though these have mostly been replaced by modern formulations.
Varieties of creosote have also been made from both oil shale and petroleum, and are known as oil-tar creosote when derived from oil tar, and as water-gas-tar creosote when derived from the tar of water gas. Creosote also has been made from pre-coal formations such as lignite, yielding lignite-tar creosote, and peat, yielding peat-tar creosote.
Creosote oils
The term creosote has a broad range of definitions depending on the origin of the coal tar oil and end-use of the material.
With respect to wood preservatives, the United States Environmental Protection Agency (EPA) considers the term creosote to mean a pesticide for use as a wood preservative meeting the American Wood Protection Association (AWPA) Standards P1/P13 and P2.[6] The AWPA Standards require that creosote "shall be a pure coal tar product derived entirely from tar produced by the carbonization of bituminous coal."[7][8]
Currently, all creosote-treated wood products—foundation and marine pilings, lumber, posts, railroad crossties, timbers, and utility poles—are manufactured using this type of wood preservative. The manufacturing process can only be a pressure process under the supervision of a licensed applicator certified by the State Departments of Agriculture. No brush-on, spray, or non-pressure uses of creosote are allowed, as specified by the EPA approved label for the use of creosote.[7]
The use of creosote according to the AWPA Standards does not allow for mixing with other types of "creosote type" materials—such as lignite-tar creosote, oil-tar creosote, peat-tar creosote, water-gas-tar creosote, or wood-tar creosote. The AWPA Standard P3 does however, allow blending of a high-boiling petroleum oil meeting the AWPA Standard P4.[8][9]
The information that follows describing the other various types of creosote materials and its uses should be considered as primarily being of only historical value. This history is important, because it traces the origin of these different materials used during the 19th and early 20th centuries. Furthermore, it must be considered that these other types of creosotes – lignite-tar, wood-tar, water-gas-tar, etc. – are not currently being manufactured and have either been replaced with more-economical materials, or replaced by products that are more efficacious or safer.
For some part of their history, coal-tar creosote and wood-tar creosote were thought to have been equivalent substances—albeit of distinct origins—accounting for their common name; the two were determined only later to be chemically different. All types of creosote are composed of phenol derivatives and share some quantity of monosubstituted phenols,[10] but these are not the only active element of creosote. For their useful effects, coal-tar creosote relies on the presence of naphthalenes and anthracenes, while wood-tar creosote relies on the presence of methyl ethers of phenol. Otherwise, either type of tar would dissolve in water.
Creosote was first discovered in its wood-tar form in 1832, by Carl Reichenbach, when he found it both in the tar and in pyroligneous acids obtained by a dry distillation of beechwood. Because pyroligneous acid was known as an antiseptic and meat preservative, Reichenbach conducted experiments by dipping meat in a dilute solution of distilled creosote. He found that the meat was dried without undergoing putrefaction and had attained a smoky flavor.[11] This led him to reason that creosote was the antiseptic component contained in smoke, and he further argued that the creosote he had found in wood tar was also in coal tar, as well as amber tar and animal tar, in the same abundance as in wood tar.[3]
Soon afterward, in 1834, Friedrich Ferdinand Runge discovered carbolic acid (phenol) in coal-tar, and Auguste Laurent obtained it from "phenylhydrate", which was soon determined to be the same compound. There was no clear view on the relationship between carbolic acid and creosote; Runge described it as having similar caustic and antiseptic properties, but noted that it was different, in that it was an acid and formed salts. Nonetheless, Reichenbach argued that creosote was also the active element, as it was in pyroligneous acid. Despite evidence to the contrary, his view held sway with most chemists, and it became commonly accepted wisdom that creosote, carbolic acid, and phenylhydrate were identical substances, with different degrees of purity.[3]
Carbolic acid was soon commonly sold under the name "creosote", and the scarcity of wood-tar creosote in some places led chemists to believe that it was the same substance as that described by Reichenbach. In the 1840s, Eugen Freiherr von Gorup-Besanez, after realizing that two samples of substances labelled as creosote were different, started a series of investigations to determine the chemical nature of carbolic acid, leading to a conclusion that it more resembled chlorinated quinones and must have been a different, entirely unrelated substance.
Independently, there were investigations into the chemical nature of creosote. A study by F.K. Völkel revealed that the smell of purified creosote resembled that of guaiacol, and later studies by Heinrich Hlasiwetz identified a substance common to guaiacum and creosote that he called creosol, and he determined that creosote contained a mixture of creosol and guaiacol. Later investigations by Gorup-Besanez, A.E. Hoffmann, and Siegfried Marasse showed that wood-tar creosote also contained phenols, giving it a feature in common with coal-tar creosote.[12]
Historically, coal-tar creosote has been distinguished from what was thought of as creosote proper—the original substance of Reichenbach's discovery—and it has been referred to specifically as "creosote oil". But, because creosote from coal-tar and wood-tar are obtained from a similar process and have some common uses, they have also been placed in the same class of substances, with the terms "creosote" or "creosote oil" referring to either product.[2]
Wood-tar creosote
| ||||||||||||||||||||||||||||||
Wood-tar creosote is a colourless to yellowish greasy liquid with a smoky odor, produces a sooty flame when burned, and has a burned taste. It is non-buoyant in water, with a specific gravity of 1.037 to 1.087, retains fluidity at a very low temperature, and boils at 205-225 °C. In its purest form, it is transparent. Dissolution in water requires up to 200 times the amount of water as the base creosote.[16] This creosote is a combination of natural phenols: primarily guaiacol and creosol (4-methylguaiacol), which will typically constitute 50% of the oil; second in prevalence are cresol and xylenol; the rest being a combination of monophenols and polyphenols.
|
The simple phenols are not the only active element in wood-tar creosote. In solution, they coagulate albumin, which is a water-soluble protein found in meat, so they serve as a preserving agent, but also cause denaturation. Most of the phenols in the creosote are methoxy derivatives: they contain the methoxy group linked to the benzene nucleus (O–CH3). The high level of methyl derivates created from the action of heat on wood (also apparent in the methyl alcohol produced through distillation) make wood-tar creosote substantially different from coal-tar creosote. Guaiacol is a methyl ether of pyrocatechin, while creosol is a methyl ether of methyl-pyrocatechin, the next homolog of pyrocatechin. Methyl ethers differ from simple phenols in being less hydrophilic, caustic, and poisonous.[18] This allows meat to be successfully preserved without tissue denaturation, and allows creosote to be used as a medical ointment.[19]
|

Because wood-tar creosote is used for its guaiacol and creosol content, it is generally derived from beechwood rather than other woods, since it distills with a higher proportion of those chemicals to other phenolics. The creosote can be obtained by distilling the wood tar and treating the fraction heavier than water with a sodium hydroxide solution. The alkaline solution is then separated from the insoluble oily layer, boiled in contact with air to reduce impurities, and decomposed by diluted sulphuric acid. This produces a crude creosote, which is purified by re-solution in alkali, re-precipitation with acid, then redistilled with the fraction passing over between 200° and 225° constituting the purified creosote.[21]
When ferric chloride is added to a dilute solution, it will turn green: a characteristic of ortho-oxy derivatives of benzene.[18] It dissolves in sulphuric acid to a red liquid, which slowly changes to purple-violet. Shaken with hydrochloric acid in the absence of air, it becomes red, the color changing in the presence of air to dark brown or black.[19]
In preparation of food by smoking, guaiacol contributes mainly to the smoky taste, while the dimethyl ether of pyrogallol, syringol, is the main chemical responsible for the smoky aroma.
Industrial
Soon after it was discovered and recognized as the principle of meat smoking, wood-tar creosote became used as a replacement for the process. Several methods were used to apply the creosote. One was to dip the meat in pyroligneous acid or a water of diluted creosote, as Reichenbach did, or brush it over with them, and within one hour the meat would have the same quality of that of traditionally smoked preparations.[22] Sometimes the creosote was diluted in vinegar rather than water, as vinegar was also used as a preservative.[23] Another was to place the meat in a closed box, and place with it a few drops of creosote in a small bottle. Because of the volatility of the creosote, the atmosphere was filled with a vapour containing it, and it would cover the flesh.[22]
The application of wood tar to seagoing vessels was practiced through the 18th century and early 19th century, before the creosote was isolated as a compound. Wood-tar creosote was found not to be as effective in wood treatments, because it was harder to infuse the creosote into the wood cells, but still experiments[24] were done, including by many governments, because it proved to be less expensive on the market.[25]
Medical
Even before creosote as a chemical compound was discovered, it was the chief active component of medicinal remedies in different cultures around the world.
In antiquity, pitches and resins were used commonly as medicines. Pliny mentions a variety of tar-like substances being used as medicine, including cedria and pissinum.[26] Cedria was the pitch and resin of the cedar tree, being equivalent to the oil of tar and pyroligneous acid which are used in the first stage of distilling creosote.[27][28] He recommends cedria to ease the pain in a toothache, as an injection in the ear in case of hardness of hearing, to kill parasitic worms, as a preventive for infusion, as a treatment for phthiriasis and porrigo, as an antidote for the poison of the sea hare, as a liniment for elephantiasis, and as an ointment to treat ulcers both on the skin and in the lungs.[28] He further speaks of cedria being used as the embalming agent for preparing mummies.[26] Pissinum was a tar water that was made by boiling cedria, spreading wool fleeces over the vessels to catch the steam, and then wringing them out.[29][30]

The Pharmacopée de Lyon, published in 1778, says that cedar tree oil is believed to cure vomiting and help medicate tumors and ulcers.[31][32] Physicians contemporary to the discovery of creosote recommended ointments and pills made from tar or pitch to treat skin diseases.[31] Tar water had been used as a folk remedy since the Middle Ages to treat affections like dyspepsia. Bishop Berkeley wrote several works on the medical virtues of tar water, including a philosophical work in 1744 titled Siris: a chain of philosophical reflexions and inquiries concerning the virtues of tar water, and divers other subjects connected together and arising one from another, and a poem where he praised its virtues.[33] Pyroligneous acid was also used at the time in a medicinal water called Aqua Binelli.[31]
Given this history, and the antiseptic properties known to creosote, it became popular among physicians in the 19th century. A dilution of creosote in water was sold in pharmacies as Aqua creosoti, as suggested by the previous use of pyroligneous acid. It was prescribed to quell the irritability of the stomach and bowels and detoxify, treat ulcers and abscesses, neutralize bad odors, and stimulate the mucous tissues of the mouth and throat.[34][35] Creosote in general was listed as an irritant, styptic, antiseptic, narcotic, and diuretic, and in small doses when taken internally as a sedative and anaesthetic. It was used to treat ulcers, and as a way to sterilize the tooth and deaden the pain in case of a tooth-ache.[34]
Creosote was suggested as a treatment for tuberculosis by Reichenbach as early as 1833. Following Reichenbach, it was argued for by John Elliotson and Sir John Rose Cormack.[34] Elliotson, inspired by the use of creosote to arrest vomiting during an outbreak of cholera, suggested its use for tuberculosis through inhalation. He also suggested it for epilepsy, neuralgia, diabetes, and chronic glanders.[36] The idea of using it for tuberculosis failed to be accepted. Use for this purpose was dropped, until the idea was revived in 1876 by British doctor G. Anderson Imlay, who suggested it be applied locally by spray to the bronchial mucous membrane.[34][37][38] This was followed up in 1877 when it was argued for in a clinical paper by Charles Bouchard and Henri Gimbert.[39] Germ theory had been established by Pasteur in 1860, and Bouchard, arguing that a bacillus was responsible for the disease, sought to rehabilitate creosote for its use as an antiseptic to treat it. He began a series of trials with Gimbert to convince the scientific community, and claimed a promising cure rate.[40] A number of publications in Germany confirmed his results in the following years.[39]
Later, a period of experimentation with different techniques and chemicals using creosote in treating tuberculosis lasted until about 1910, when radiation therapy seemed more promising. Guaiacol, instead of a full creosote solution, was suggested by Hermann Sahli in 1887. He argued it had the active chemical of creosote and had the advantage of being of definite composition and having a less unpleasant taste and odor.[41] A number of solutions of both creosote and guaiacol appeared on the market, such as phosphotal and guaicophosphal, phosphites of creosote and guaiacol; eosot and geosot, valerinates of creosote and guaicol; phosot and taphosot, phosphate and tannophospate of creosote; and creosotal and tanosal, tannates of creosote.[42] Creosote and eucalyptus oil were also a remedy used together, administered through a vaporizor and inhaler. Since then, more effective and safer treatments for tuberculosis have been developed.
In the 1940s, Canadian-based Eldon Boyd experimented with guaiacol and a recent synthetic modification—glycerol guaiacolate (guaifenesin)—on animals. His data showed that both drugs were effective in increasing secretions into the airways in laboratory animals, when high enough doses were given.
Industrial
Wood-tar creosote is to some extent used for wood preservation, but it is generally mixed with coal-tar creosote, since the former is not as effective. Commercially available preparations of "liquid smoke", marketed to add a smoked flavour to meat and aid as a preservative, consist primarily of creosote and other constituents of smoke.[43] Creosote is the ingredient that gives liquid smoke its function; guaicol lends to the taste and the creosote oils help act as the preservative. Creosote can be destroyed by treatment with chlorine, either sodium hypochlorite, or calcium hypochlorite solutions. The phenol ring is essentially opened, and the molecule is then subject to normal digestion and normal respiration.
Medical
The guaifenesin developed by Eldon Boyd is still commonly used today as an expectorant, sold over the counter, and usually taken by mouth to assist the bringing up of phlegm from the airways in acute respiratory tract infections. Guaifenesin is a component of Mucinex, Robitussin DAC, Cheratussin DAC, Robitussin AC, Cheratussin AC, Benylin, DayQuil Mucous Control, Meltus, and Bidex 400.
Seirogan is a popular Kampo medicine in Japan, used as an anti-diarrheal, and has 133 mg wood creosote from beech, pine, maple or oak wood per adult dose as its primary ingredient. Seirogan was first used as a gastrointestinal medication by the Imperial Japanese Army in Russia during the Russo-Japanese War of 1904 to 1905.[44]
Creomulsion is a cough medicine in the United States, introduced in 1925, that is still sold and contains beechwood creosote. Beechwood creosote is also found under the name kreosotum or kreosote.
Coal-tar creosote
|
Coal-tar creosote is greenish-brown liquid, with different degrees of darkness, viscosity, and fluorescence depending on how it is made. When freshly made, the creosote is a yellow oil with a greenish cast and highly fluorescent, and the fluorescence is increased by exposure to air and light. After settling, the oil is dark green by reflected light and dark red by transmitted light.[47] To the naked eye, it generally appears brown. The creosote (often called "creosote oil") consists almost wholly of aromatic hydrocarbons, with some amount of bases and acids and other neutral oils. The flash point is 70–75 °C and burning point is 90–100 °C,[48] and when burned it releases a greenish smoke.[49] The smell largely depends on the naphtha content in the creosote. If there is a high amount, it will have a naphtha-like smell, otherwise it will smell more of tar.
In the process of coal-tar distillation, the distillate is collected into four fractions; the "light oil", which remains lighter than water, the "middle oil" which passes over when the light oil is removed; the "heavy oil", which sinks; and the "anthracene oil", which when cold is mostly solid and greasy, of a buttery consistence. Creosote refers to the portion of coal tar which distills as "heavy oil", typically between 230 and 270 °C, also called "dead oil"; it sinks into water but still is fairly liquid. Carbolic acid is produced in the second fraction of distillation and is often distilled into what is referred to as "carbolic oil".[50][51][52][53]
|

Commercial creosote will contain substances from six groups.[45] The two groups occur in the greatest amounts and are the products of the distillation process—the "tar acids", which distill below 205 °C and consist mainly of phenols, cresols, and xylenols, including carbolic acid—and aromatic hydrocarbons, which divide into naphthalenes, which distill approximately between 205 and 255 °C, and constituents of an anthracene nature, which distill above 255 °C.[55] The quantity of each varies based on the quality of tar and temperatures used, but generally, the tar acids won't exceed 5%, the naphthalenes will make up 15 to 50%, and the anthracenes will make up 45% to 70%.[55] The hydrocarbons are mainly aromatic; derivatives of benzene and related cyclic compounds such as naphthalene, anthracene, phenanthrene, acenaphthene, and fluorene. Creosotes from vertical-retort and low temperature tars contain, in addition, some paraffinic and olefinic hydrocarbons. The tar-acid content also depends on the source of the tar—it may be less than 3% in creosote from coke-oven tar and as high as 32% in creosote from vertical retort tar.[56] All of these have antiseptic properties. The tar acids are the strongest antiseptics but have the highest degree of solubility in water and are the most volatile; so, like with wood-tar creosote, phenols are not the most valued component, as by themselves they would lend to being poor preservatives.[57] In addition, creosote will contain several products naturally occurring in coal—nitrogen-containing heterocycles, such as acridines, carbazoles, and quinolines, referred to as the "tar bases" and generally make up about 3% of the creosote—sulfur-containing heterocycles, generally benzothiophenes[58]—and oxygen-containing heterocycles, dibenzofurans.[59] Lastly, creosote will contain a small number of aromatic amines produced by the other substances during the distillation process and likely resulting from a combination of thermolysis and hydrogenation.[60][61] The tar bases are often extracted by washing the creosote with aqueous mineral acid,[56] although they're also suggested to have antiseptic ability similar to the tar acids.
Commercially used creosote is often treated to extract the carbolic acid, naphthalene, or anthracene content. The carbolic acid or naphthalene is generally extracted to be used in other commercial products.[62] American produced creosote oils typically will have low amounts of anthracene and high amounts of naphthalene, because when forcing the distillate at a temperature that produces anthracene the soft pitch will be ruined and only the hard pitch will remain; this ruins it for use in roofing purposes, and only leaves a product which isn't commercially useful.[61]
Industrial
The use of coal-tar creosote on a commercial scale began in 1838, when a patent covering the use of creosote oil to treat timber was taken out by inventor John Bethell. The "Bethell process"—or as it later became known, the full-cell process—involves placing wood to be treated in a sealed chamber and applying a vacuum to remove air and moisture from wood "cells". The wood is then pressure-treated to imbue it with creosote or other preservative chemicals, after which vacuum is reapplied to separate the excess treatment chemicals from the timber. Alongside the zinc chloride-based "Burnett process", use of creosoted wood prepared by the Bethell process became a principal way of preserving railway timbers (most notably railway sleepers) to increase the lifespan of the timbers, and avoiding having to regularly replace them.[63]
Besides treating wood, it was also used for lighting and fuel. In the beginning, it was only used for lighting needed in harbour and outdoor work, where the smoke that was produced from burning it was of little inconvenience. By 1879, lamps had been created that ensured a more complete combustion by using compressed air, removing the drawback of the smoke. Creosote was also processed into gas and used for lighting that way. As a fuel, it was used to power ships at sea and blast furnaces for different industrial needs, once it was discovered to be more efficient than unrefined coal or wood. It was also used industrially for the softening of hard pitch, and burned to produce lamp black. By 1890, the production of creosote in the United Kingdom totaled approximately 29,900,000 gallons per year.[49]
In 1854, Alexander McDougall and Robert Angus Smith developed and patented a product called McDougall's Powder as a sewer deodorant; it mainly consisted of carbolic acid derived from creosote. McDougall, in 1864, experimented with his solution to remove entozoa parasites from cattle pasturing on a sewage farm.[64] This later led to widespread use of creosote as a cattle wash and sheep dip. External parasites would be killed in a creosote diluted dip, and drenching tubes would be used to administer doses to the animals' stomachs to kill internal parasites.[65]
Two later methods for creosoting wood were introduced after the turn of the century, referred to as empty-cell processes, because they involve compressing the air inside the wood so that the preservative can only coat the inner cell walls rather than saturating the interior cell voids. This is a less effective, though usually satisfactory, method of treating the wood, but is used because it requires less of the creosoting material. The first method, the "Rüping process" was patented in 1902, and the second, the "Lowry process" was patented in 1906. Later in 1906, the "Allardyce process" and "Card process" were patented to treat wood with a combination of both creosote and zinc chloride.[63] In 1912, it was estimated that a total of 150,000,000 gallons were produced in the US per year.
Medical
Coal-tar creosote, despite its toxicity, was used as a stimulant and escharotic, as a caustic agent used to treat ulcers and malignancies and cauterize wounds and prevent infection and decay. It was particularly used in dentistry to destroy tissues and arrest necrosis.[66][67][68]
Industrial
Coal-tar creosote is the most widely used wood treatment today; both industrially, processed into wood using pressure methods such as "full-cell process" or "empty-cell process", and more commonly applied to wood through brushing. In addition to toxicity to fungi, insects, and marine borers, it serves as a natural water repellent. It is commonly used to preserve and waterproof cross ties, pilings, telephone poles, power line poles, marine pilings, and fence posts. Although suitable for use in preserving the structural timbers of buildings, it is not generally used that way because it is difficult to apply. There are also concerns about the environmental impact of the leaching of creosote into aquatic ecosystems.
Due to its carcinogenic character, the European Union has regulated the quality of creosote for the EU market[69] and requires that the sale of creosote be limited to professional users.[70][71] The United States Environmental Protection Agency regulates the use of coal-tar creosote as a wood preservative under the provisions of the Federal Insecticide, Fungicide, and Rodenticide Act. Creosote is considered a restricted-use pesticide and is only available to licensed pesticide applicators.[72][73]
Oil-tar creosote
|

Oil-tar creosote is derived from the tar that forms when using petroleum or shale oil in the manufacturing of gas. The distillation of the tar from the oil occurs at very high temperatures; around 980 °C. The tar forms at the same time as the gas, and once processed for creosotes contains a high percentage of cyclic hydrocarbons, a very low amount of tar acids and tar bases, and no true anthracenes have been identified.[74] Historically, this has mainly been produced in the United States on the Pacific coast, where petroleum has been more abundant than coal. Limited quantities have been used industrially, either alone, mixed with coal-tar creosote, or fortified with pentachlorophenol.[75]
Water-gas-tar creosote
Water-gas-tar creosote is also derived from petroleum oil or shale oil, but by a different process; it is distilled during the production of water gas. The tar is a by-product resulting from enrichment of water gas with gases produced by thermal decomposition of petroleum. Of the creosotes derived from oil, it is practically the only one used for wood preservation. It has the same degree of solubility as coal-tar creosote and is easy to infuse into wood. Like standard oil-tar creosote, it has a low amount of tar acids and tar bases, and has less antiseptic qualities.[54] Petri dish tests have shown that water-gas-tar creosote is one-sixth as anti-septically effective as that of coal-tar.[76]
Lignite-tar creosote
Lignite-tar creosote is produced from lignite rather than bituminous coal, and varies considerably from coal-tar creosote. Also called "lignite oil", it has a very high content of tar acids, and has been used to increase the tar acids in normal creosote when necessary.[77] When it has been produced, it has generally been applied in mixtures with coal-tar creosote or petroleum. Its effectiveness when used alone has not been established. In an experiment with southern yellow pine fence posts in Mississippi, straight lignite-tar creosote was giving good results after about 27 years exposure, although not as good as the standard coal-tar creosote used in the same situation.[78]
Health effects
According to the Agency for Toxic Substances and Disease Registry (ATSDR), eating food or drinking water contaminated with high levels of coal-tar creosote may cause a burning in the mouth and throat, and stomach pains. ATSDR also states that brief direct contact with large amounts of coal-tar creosote may result in a rash or severe irritation of the skin, chemical burns of the surfaces of the eyes, convulsions and mental confusion, kidney or liver problems, unconsciousness, and even death. Longer direct skin contact with low levels of creosote mixtures or their vapours can result in increased light sensitivity, damage to the cornea, and skin damage. Longer exposure to creosote vapours can cause irritation of the respiratory tract.
The International Agency for Research on Cancer (IARC) has determined that coal-tar creosote is probably carcinogenic to humans, based on adequate animal evidence and limited human evidence. It is instructive to note that the animal testing relied upon by IARC involved the continuous application of creosote to the shaved skin of rodents. After weeks of creosote application, the animals developed cancerous skin lesions and in one test, lesions of the lung. The United States Environmental Protection Agency has stated that coal-tar creosote is a probable human carcinogen based on both human and animal studies.[80] As a result, the Federal Occupational Safety and Health Administration (OSHA) has set a permissible exposure limit of 0.2 milligrams of coal-tar creosote per cubic meter of air (0.2 mg/m3) in the workplace during an 8-hour day, and the Environmental Protection Agency (EPA) requires that spills or accidental releases into the environment of one pound (0.454 kg) or more of creosote be reported to them.
There is no unique exposure pathway of children to creosote. Children exposed to creosote will probably experience the same health effects seen in adults exposed to creosote. It is unknown whether children differ from adults in their susceptibility to health effects from creosote.
A 2005 mortality study of creosote workers found no evidence supporting an increased risk of cancer death, as a result of exposure to creosote. Based on the findings of the largest mortality study to date of workers employed in creosote wood treating plants, there is no evidence that employment at creosote wood-treating plants or exposure to creosote-based preservatives was associated with any significant mortality increase from either site-specific cancers or non-malignant diseases. The study consisted of 2,179 employees at eleven plants in the United States where wood was treated with creosote preservatives. Some workers began work in the 1940s to 1950s. The observation period of the study covered 1979–2001. The average length of employment was 12.5 years. One third of the study subjects were employed for over 15 years.[81]
The largest health effect of creosote is deaths caused by residential chimney fires due to chimney tar (creosote) build-up. This is entirely unconnected with its industrial production or use.[82]
Build-up in chimneys
Burning wood and fossil fuels in the absence of adequate airflow (such as in an enclosed furnace or stove), causes incomplete combustion of the oils in the wood, which are off-gassed as volatiles in the smoke. As the smoke rises through the chimney it cools, causing water, carbon, and volatiles to condense on the interior surfaces of the chimney flue. The black oily residue that builds up is referred to as creosote, which is similar in composition to the commercial products by the same name, but with a higher content of carbon black.
Over the course of a season creosote deposits can become several inches thick. This creates a compounding problem, because the creosote deposits reduce the draft (airflow through the chimney) which increases the probability that the wood fire is not getting enough air for complete combustion. Since creosote is highly combustible, a thick accumulation creates a fire hazard. If a hot fire is built in the stove or fireplace, and the air control left wide open, this may allow hot oxygen into the chimney where it comes in contact with the creosote which then ignites—causing a chimney fire. Chimney fires often spread to the main building because the chimney gets so hot that it ignites any combustible material in direct contact with it, such as wood. The fire can also spread to the main building from sparks emitting from the chimney and landing on combustible roof surfaces. In order to properly maintain chimneys and heaters that burn wood or carbon-based fuels, the creosote buildup must be removed. Chimney sweeps perform this service for a fee.[82]
Release into environment

Even though creosote is pressurized into the wood, the release of the chemical – and resulting marine pollution – occurs due to many different events: During the lifetime of the marine piling, weathering occurs from tides and water flow which slowly opens the oily outer coating and exposes the smaller internal pores to more water flow.[83] Frequent weathering occurs daily, but more severe weather, such as hurricanes, can cause damage or loosening of the wooden pilings.[83] Many pilings are either broken into pieces from debris, or are completely washed away during these storms. When the pilings are washed away, they come to settle on the bottom of the body of water where they reside, and then they leach chemicals into the water slowly over a long period of time. This long-term secretion is not normally noticed because the piling is submerged beneath the surface, hidden from sight.
The creosote is mostly insoluble in water, but the lower-molecular-weight compounds will become soluble the longer the broken wood is exposed to the water.[84] In this case, some of the chemicals become water-soluble and further leach into the aquatic sediment while the rest of the insoluble chemicals remain together in a tar-like substance.[84] Another source of damage comes from wood-boring fauna, such as shipworms and Limnoria.[85] Though creosote is used as a pesticide preservative, studies have shown that Limnoria is resistant to wood preservative pesticides and can cause small holes in the wood, through which creosote can then be released.[85]
Chemical reactions with sediment and organisms
Once the soluble compounds from the creosote preservative leach into the water, the compounds begin reacting with the external environment or are consumed by organisms. The reactions vary depending on the concentration of each compound that is released from the creosote, but major reactions are outlined below:
Alkylation
Alkylation occurs when a molecule replaces a hydrogen atom with an alkyl group that generally comes from an organic molecule.[86] Alkyl groups that are found naturally occurring in the environment are organometallic compounds.[87] Organometallic compounds generally contain a methyl, ethyl, or butyl derivative which is the alkyl group that replaces the hydrogen.[87] Other organic compounds, such as methanol, can provide alkyl groups for alkylation.[88] Methanol is found naturally in the environment in small concentrations, and has been linked to the release from biological decomposition of waste and even a byproduct of vegetation.[89] The following reactions are alkylations of soluble compounds found in creosote preservatives with methanol.
m-Cresol
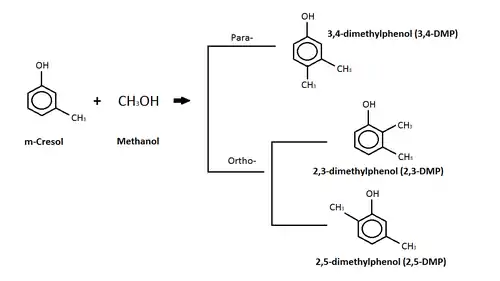
The diagram above depicts a reaction between m-cresol and methanol where a c-alkylation product is produced.[88] The c-alkylation reaction means that instead of replacing the hydrogen atom on the -OH group, the methyl group (from the methanol) replaces the hydrogen on a carbon in the benzene ring.[88] The products of this c-alkylation can be in either a para- or ortho- orientation on the molecule, as seen in the diagram, and water, which is not shown.[88] Isomers of the dimethylphenol (DMP) compound are the products of the para- and ortho-c-alkylation.[88] Dimethylphenol (DMP) compound is listed as an aquatic hazard by characteristic, and is toxic with long lasting effects.[90]
Phenol
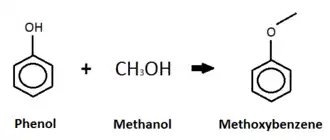
This diagram shows an o-alkylation between phenol and methanol. Unlike the c-alkylation, the o-alkylation replaces the hydrogen atom on the -OH group with the methyl group (from the methanol).[91] The product of the o-alkylation is methoxybenzene, better-known as anisole, and water, which is not shown in the diagram.[91] Anisole is listed as an acute hazard to aquatic life with long-term effects.[92]
Bioaccumulation
Bioaccumulation is the process by which an organism takes in chemicals through ingestion, exposure, and inhalation.[93] Bioaccumulation is broken down into bioconcentration (uptake of chemicals from the environment) and biomagnification (increasing concentration of chemicals as they move up the food chain).[93] Certain species of aquatic organisms are affected differently from the chemicals released from creosote preservatives. One of the more studied organisms is a mollusk. Mollusks attach to the wooden, marine pilings and are in direct contact with the creosote preservatives.[94] Many studies have been conducted using polycyclic aromatic hydrocarbons (PAH), which are low molecular hydrocarbons found in some creosote-based preservatives. In a study conducted from Pensacola, Florida, a group of native mollusks were kept in a controlled environment, and a different group of native mollusks were kept in an environment contaminated with creosote preservatives.[95] The mollusks in the contaminated environment were shown to have a bioaccumulation of up to ten times the concentration of PAH than the control species.[95] The intake of organisms is dependent on whether the compound is in an ionized or an un-ionized form.[96] To determine whether the compound is ionized or un-ionized, the pH of the surrounding environment must be compared to the pKa or acidity constant of the compound.[96] If the pH of the environment is lower than the pKa, then the compound is un-ionized which means that the compound will behave as if it is non-polar.[96] Bioaccumulation for un-ionized compounds comes from partitioning equilibrium between the aqueous phase and the lipids in the organism.[96] If the pH is higher than the pKa, then the compound is considered to be in the ionized form.[96] The un-ionized form is favored because the bioaccumulation is easier for the organism to intake through partitioning equilibrium.[96] The table below shows a list of pKas from compounds found in creosote preservatives and compares them to the average pH of seawater (reported to be 8.1).[97]
| Compound | pKa | pH of Seawater | Form (Ionized or Un-Ionized) |
|---|---|---|---|
| m-cresol | 10.09 | 8.1 | Un-ionized |
| o-cresol | 10.29 | Un-ionized | |
| p-cresol | 10.30 | Un-ionized | |
| 2-ethylphenol | 10.20 | Un-ionized | |
| guaiacol | 9.98 | Un-ionized | |
| phenol | 9.99 | Un-ionized |
Each of the compounds in the table above is found in creosote preservatives; all are in the favored un-ionized form. In another study, various species of small fish were tested to see how the exposure time to PAH chemicals affected the fish.[7] This study showed that an exposure time of 24–96 hours on various shrimp and fish species affected the growth, reproduction, and survival functions of the organisms for most of the compounds tested.[7]
Biodegradation
Biodegradation can be seen in some studies that biodegradation accounts for the absence of creosote preservatives on the initial surface of the sediment.[95] In a study from Pensacola, Florida, PAHs were not detected on the surface on the aquatic sediment, but the highest concentrations were detected at a depth of 8-13 centimeters.[95] A form an anaerobic biodegradation of m-cresol was seen in a study using sulfate-reducing and nitrate-reducing enriched environments.[98] The reduction of m-cresol in this study was seen in under 144 hours, while additional chemical intermediates were being formed.[98] The chemical intermediates were formed in the presence of bicarbonate. The products included 4-hydroxy-2-methylbenzoic acid and acetate compounds.[98] Although the conditions were enriched with the reducing anaerobic compounds, sulfate and nitrate reducing bacteria are commonly found in the environment. For further information, see sulfate-reducing bacteria. The type of anaerobic bacteria ultimately determines the reduction of the creosote preservative compounds, while each individual compound may only go through reduction under certain conditions.[99] BTEX is a mixture of benzene, toluene, ethylbenzene, and xylene, that was studied in the presence of four different anaerobic-enriched sediments.[99] Though the compound, BTEX, is not found in creosote preservatives, the products of creosote preservatives' oxidation-reduction reactions include some of these compounds. For oxidation-reduction reactions, see the following section. In this study, it was seen that certain compounds such as benzene were only reduced under sulfate-enriched environments, while toluene was reduced under a variety of bacteria-enriched environments, not just sulfate.[99] The biodegradation of a creosote preservative in an anaerobic enrichment depends not only on the type of bacteria enriching the environment, but also the compound that has been released from the preservative. In aerobic environments, preservative compounds are limited in the biodegradation process by the presence of free oxygen.[100] In an aerobic environment, free oxygen comes from oxygen saturated sediments, sources of precipitation, and plume edges.[100] The free oxygen allows for the compounds to be oxidized and decomposed into new intermediate compounds.[100] Studies have shown that when BTEX and PAH compounds were placed in aerobic environments, the oxidation of the ring structures caused cleavage in the aromatic ring and allowed for other functional groups to attach.[100] When an aromatic hydrocarbon was introduced to the molecular oxygen in experimental conditions, a dihydrodiol intermediate was formed, and then oxidation occurred transforming the aromatic into a catechol compound.[100] Catechol allows for cleavage of the aromatic ring to occur, where functional groups can then add in an ortho- or meta- position.[100]
Oxidation-reduction
Even though many studies conduct testing under experimental or enriched conditions, oxidation-reduction reactions occur naturally and allow for chemicals to go through processes such as biodegradation, outlined above. Oxidation is defined as the loss of an electron to another species, while reduction is the gaining of an electron from another species. As compounds go through oxidation and reduction in sediments, the preservative compounds are altered to form new chemicals, leading to decomposition. An example of the oxidation of p-cresol and phenol can be seen in the figures below:
p-Cresol
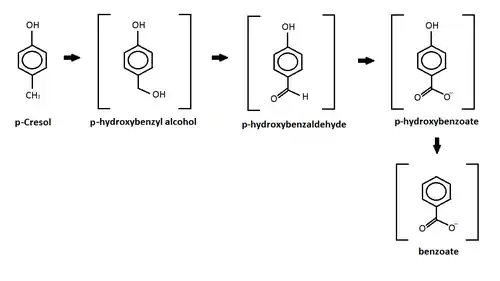
This reaction shows the oxidation of p-cresol in a sulfate-enriched environment.[101] P-cresol was seen to be the easiest to degrade through the sulfate-enriched environment, while m-cresol and o-cresol where inhibited.[101] In the chart above, p-cresol was oxidized under an anaerobic sulfate reducing condition and formed four different intermediates.[101] After the formation of the intermediates, the study reported further degradation of the intermediates leading to the production of carbon dioxide and methane.[101] The p-hydroxylbenzyl alcohol, p-hydroxylbenzaldehye, p-hyrdoxylbenzoate, and benzoate intermediates all are produced from this oxidation and released into the sediments.[101] Similar results were also produced by different studies using other forms of oxidation such as: iron-reducing organisms,[102] Copper/Manganese Oxide catalyst,[103] and nitrate- reducing conditions.[104]
Phenol
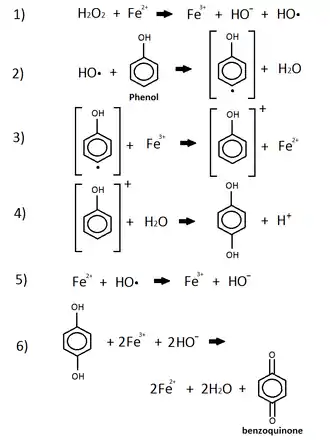
This reaction shows the oxidation of phenol by iron and peroxide.[105] This combination of iron, which comes from iron oxide in the sediment, and the peroxide, commonly released by animals and plants into the environment, is known as the Fenton Reagent.[105] This reagent is used to oxidize phenol groups by the use of a radical hydroxide group produced from the peroxide in the p-benzoquinone.[105] This product of phenol's oxidation is now leached into the environment while other products include iron(II) and water. P-benzoquinone is listed as being a very toxic, acute environmental hazard.[106]
Environmental hazards
Sediment
In aquatic sediments, several reactions can transform the chemicals released by the creosote preservatives into more dangerous chemicals. Most creosote preservative compounds have hazards associated with them before they are transformed. Cresol (m-, p-, and o-), phenol, guaiacol, and xylenol (1,3,4- and 1,3,5-) all are acute aquatic hazards prior to going through chemical reactions with the sediments. Alkylation reactions allows for the compounds to transition into more toxic compounds with the addition of R-groups to the major compounds found in creosote preservatives. Compounds formed through alkylation include: 3,4-dimethylphenol, 2,3-dimethylphenol, and 2,5-dimethylphenol, which are all listed as acute environmental hazards.[88] Biodegradation controls the rate at which the sediment holds the chemicals, and the number of reactions that are able to take place. The biodegradation process can take place under many different conditions, and vary depending on the compounds that are released. Oxidation-reduction reactions allow for the compounds to be broken down into new forms of more toxic molecules. Studies have shown oxidation-reduction reactions of creosote preservative compounds included compounds that are listed as environmental hazards, such as p-benzoquinone in the oxidation of phenol.[105] Not only are the initial compounds in creosote hazardous to the environment, but the byproducts of the chemical reactions are environmental hazardous as well.
Other
From the contamination of the sediment, more of the ecosystem is affected. Organisms in the sediment are now exposed to the new chemicals. Organisms are then ingested by fish and other aquatic animals. These animals now contain concentrations of hazardous chemicals which were secreted from the creosote. Other issues with ecosystems include bioaccumulation. Bioaccumulation occurs when high levels of chemicals are passed to aquatic life near the creosote pilings. Mollusks and other smaller crustaceans are at higher risk because they are directly attached to the surface of wood pilings that are filled with creosote preservative. Studies show that mollusks in these environments take on high concentrations of chemical compounds which will then be transferred through the ecosystem's food chain. Bioaccumulation contributes to the higher concentrations of chemicals within the organisms in the aquatic ecosystems.[107]
See also
- Pentachlorophenol
- Creolin
Notes
- Delnao 1943
- Price, Kelogg & Cox 1909, p. 7
- Schorlemmer 1885, p. 152
- "ATSDR - ToxFAQs™: Creosote". www.atsdr.cdc.gov. Retrieved 2020-11-24.
- "Coal Tar and Coal-Tar Pitch - Cancer-Causing Substances - National Cancer Institute". www.cancer.gov. 2015-03-20. Retrieved 2020-11-24.
- Communication between United States Environmental Protection Agency and the Creosote Council.
- "Reregistration Eligibility Decision for Creosote (Case 0139)" (PDF). United States Environmental Protection Agency. September 25, 2008. Retrieved October 29, 2016.
- 2013 AWPA Book of Standards. American Wood Protection Association.
- MacLean 1952
- Roscoe & Schorlemmer 1888, p. 37
- Roscoe & Schorlemmer 1888, p. 33
- Schorlemmer 1885, p. 153
- Allen 1910, p. 353
- American Pharmaceutical Association 1895, p. 1073
- Renard 1895, p. 294
- Nickels 1890, p. 614
- Lee et al. 2005, p. 1483
- Pharmaceutical Society of Great Britain 1898, p. 468
- Allen 1910, p. 348
- Price, Kelogg & Cox 1909, p. 13
- Allen 1910, p. 347
- Abel & Smith 1857, p. 23
- Letheby 1870, pp. 225–226
- Joerin 1909, p. 767
- Bradbury 1909, p. 107
- Cormack 1836, p. 58
- Parr 1809, p. 383
- Pliny 1856, p. 8
- Berkeley 1744, p. 9
- Pliny 1855, p. 290
- Cormack 1836, p. 50
- Vitet 1778, p. 427
- Chemist and Druggist 1889, p. 300
- King, Felter & Llyod 1905, p. 617
- Taylor 1902, p. 207
- Whittaker 1893, p. 77
- Imlay 1876, p. 514
- Dobbell 1878, p. 315
- Kinnicutt 1892, p. 514
- Contrepois 2002, p. 211
- Kinnicutt 1892, p. 515
- Coblentz 1908
- Chenoweth 1945, p. 206
- Seirogan 2011
- Melber, Kielhorn & Mangelsdorf 2004, p. 11
- Speight 1994, p. 456
- Allen 1910, p. 366
- Bateman 1922, p. 50
- Nickels 1890, p. 615
- Philips 1891, p. 255
- Martin 1913, pp. 416–419
- Nelson 1907, p. 204
- Noller 1965, p. 185
- Price, Kelogg & Cox 1909, p. 12
- Engineering and Contracting 1912, p. 531
- Greenhow 1965, p. 58
- American Railway Bridge and Building Association 1914, p. 287
- Orr & White 1990, p. 39
- Speight 1994, p. 77
- Orr & White 1990, p. 255
- Bateman 1922, p. 47
- Mushrush & Speight 1995, p. 115
- Angier 1910, p. 408
- Brock 2008, p. 91
- Salmon 1901, pp. 7–14
- Farrar 1880, pp. 412–417
- Farrar 1893, pp. 1–25
- Pease 1862
- "Commission Directive 2001/90/EC". Official Journal of the European Communities. 27 October 2001 – via eur-lex.europa.eu.
- "Commission Directive 76/769/EEC". Official Journal of the European Communities. 3 October 2007 – via eur-lex.europa.eu.
- Health and Safety Executive 2011
- Creosote Council 2011
- Ibach & Miller 2007, 14-1–14-9
- Voorhies 1940
- Hunt & Garratt 1967, p. 88
- Stimson 1914, p. 626
- Richardson 1993, p. 103
- Hunt & Garratt 1967, p. 97
- Encyclopædia Britannica 1949, p. 821
- "Creosote (CASRN 8001-58-9)". Integrated Risk Information System (IRIS). United States Environmental Protection Agency. September 7, 1988. Archived from the original on 2000-08-23.
- Wong & Harris 2005
- DHS 2006
- Shupe, Lebow & Ring 2008
- Smith 2002
- Shupe 2012
- "Alkylation". Dictionary.com. Retrieved October 29, 2016.
- Connell 2005, pp. 376–379
- Bolognini et al 2002
- Howard 1990, p. 311
- "2,3-Dimethylphenol". PubChem Database. National Center for Biotechnology Information. Retrieved April 7, 2019.
- Balsama et al 1984
- "Anisole". PubChem Database. National Center for Biotechnology Information. Retrieved April 7, 2019.
- Clarke & McFarland 1991
- Weitkamp & Bennett 2011
- Elder & Dresler 1988
- Neff 2002
- "Ocean Acidification". Pristine Seas. National Geographic. Archived from the original on 2015-08-29.
- Ramanand & Suflita 1991
- Phelps & Young 1999
- Aronson et al 1999
- Smolenski & Suflita 1987
- Lovley & Lonergan 1990
- Wang et al 2004
- Bossert & Young 1986
- Zazo et al 2005
- "Quinone". PubChem Database. National Center for Biotechnology Information. Retrieved April 7, 2019.
- "Aquatic Food Webs". Marine Life Education Resource. National Oceanic and Atmospheric Administration. February 2019. Retrieved April 8, 2019.
References
- Abel, Ambrose; Smith, Elizur Goodrich (1857). The preservation of food: From the "Aus der natur" of Abel. Press of Case, Lockwood and company.
- Allen, Alfred Henry (1910). "Creosote and Creosote oils". Allen's Commercial Organic Analysis. 3: 346–391.
- American Pharmaceutical Association (1895). "Creosote and Creosote oils". Proceedings of the American Pharmaceutical Association at the Annual Meeting. 43: 1073.
- American Railway Bridge and Building Association (1914). "Wood Preserving Creosotes: Methods of Production, Properties, Quality, Price and Quantity Consumed in the United States". Proceedings of the Annual Convention of the American Railway, Bridge and Building Association. 23: 287–288.
- Angier, F.J. (1910). "The seasoning and preservative treatment of wood ties". Railway Age Gazette. 48: 408–411.
- Aronson, D.; Citra, M.; Shuler, K.; Printup, H.; Howard, P.H. (January 27, 1999). Aerobic Biodegradation of Organic Chemicals in Environment Media: A Summary of Field and Laboratory Studies (PDF) (Report). Environment Science Center Syracuse Research Corporation. Archived from the original (PDF) on 2016-12-20.
- Balsama S, Beltrame P, Beltrame PL, Carniti P, Forni L, Zuretti G (December 14, 1984). "Alkylation of Phenol with Methanol over Zeolites". Applied Catalysis. 13 (1): 161–170. doi:10.1016/S0166-9834(00)83334-5.
- Bateman, Ernest (1922). Coal-tar and water-gas tar creosotes. Govt. print. off.
- Berkeley, George (1744). Siris: a Chain of Philosophical Reflexions and Inquiries Concerning the Virtues of Tar Water: And Divers Other Subjects Connected Together and Arising One from Another. Dublin; London: W. Innys, C. Hitch, C. Davis.
- Bernheim, Samuel (1901). La Tuberculose et la médication créosotée. Paris: Maloine.
- Bolognini M, Cavani F, Scagliarini D, Flego C, Perego C, Sabo M (July 2002). "Heterogeneous Basic Catalysts as Alternative to Homogeneous Catalysts:Reactivity of Mg/Al mixed Oxides in the Alkylation of m-Cresol with Methanol". Catalysis Today. 75 (1): 103–111. doi:10.1016/S0920-5861(02)00050-0.
- Bossert, ID; Young, LY (November 1986). "Anaerobic oxidation of p-cresol by a denitrifying bacterium". Applied and Environmental Microbiology. 52 (5): 1117–22. Bibcode:1986ApEnM..52.1117B. doi:10.1128/AEM.52.5.1117-1122.1986. PMC 239183. PMID 3789714.
- Bradbury, Robert H. (1909). "Increase in the use of wood preservatives indicates progress in wood preservation". Journal of the Franklin Institute. 168 (2): 107. doi:10.1016/s0016-0032(09)90070-9.
- Brock, William Hodson (2008). William Crookes and the commercialization of science. Ashgate Publishing, Ltd. ISBN 9780754663225.
- Chemist and Druggist (1889). "Tar Water". Chemist and Druggist. 35: 300.
- Chenoweth, Walter Winfred (1945). How to preserve food. Houghton Mifflin company.
- Clarke, Joan U.; McFarland, Victor A. (July 1991). Assessing Bioaccumulation in Aquatic Organisms Exposed to Contaminated Sediments (PDF) (Report). US Army Corps of Engineers. Retrieved October 29, 2016.
- Coblentz, Virgil (1908). The Newer Remedies …: A Reference Manual for Physicians, Pharmacists and Students. Apothecary Publishing.
- Connell, Des (July 14, 2005). Basic Concepts of Environmental Chemistry (2nd ed.). CRC Press. ISBN 9780203025383. Retrieved April 7, 2019.
- Contrepois, Alain (2002). "The Clinician, Germs and Infectious Diseases: The Example of Charles Bouchard in Paris". Medical History. 46 (2): 197–220. doi:10.1017/S0025727300069088. PMC 1044495. PMID 12024808.
- Cormack, Sir John Rose (1836). A treatise on the chemical, medicinal, and physiological properties of creosote: illustrated by experiments on the lower animals: with some considerations on the embalment of the Egyptians. Being the Harveian prize dissertation for 1836. J. Carfrae & Son.
- Creosote Council (2011). "Regulation". creosotecouncil.org/. Archived from the original on 2011-05-04.
- Delnao, Jack (March 1943). "At the Santa Fe R.R. tie plant, Albuquerque, N[ew] Mex[ico].…". Prints & Photographs Online Catalog. Library of Congress. Retrieved 16 February 2015.
- Dobbell, Horace (1878). "Carbolic acid and creosote". Annual Reports on Diseases of the Chest. 3: 315.
- Elder, JF; Dresler, PV (1988). "Accumulation and bioconcentration of polycyclic aromatic hydrocarbons in a nearshore estuarine environment near a Pensacola (Florida) creosote contamination site". Environmental Pollution. 49 (2): 117–132. doi:10.1016/0269-7491(88)90244-8. PMID 15092667. Retrieved October 29, 2016.
- Encyclopædia Britannica (1949). Encyclopædia Britannica: a new survey of universal knowledge. Vol. 21. Encyclopædia Britannica.
- Engineering and Contracting (1912). "Wood Preserving Creosotes: Methods of Production, Properties, Quality, Price and Quantity Consumed in the United States". Engineering and Contracting. 38 (13): 350–353.
- Farrar, J.N. (1880). "On the comparative value of sulphuric acid and creosote in the treatment of alveolar cavities". Annals of Anatomy and Surgery. 2: 412–418.
- Farrar, J.N. (1893). "Pulpless teeth; abscess; treatment, especially surgical treatment". Transactions of the New York Ondontological Society: 1–25.
- Greenhow, E.J. (1965). Wood. Vol. 30. Tothill Press.
- Hartnik T, Norli HR, Eggen T, Breedveld GD (January 2007). "Bioassay-directed identification of toxic organic compounds in creosote-contaminated groundwater". Chemosphere. 66 (3): 435–443. Bibcode:2007Chmsp..66..435H. doi:10.1016/j.chemosphere.2006.06.031. PMID 16872665.
- Health and Safety Executive (2011). "Revocation of approvals for amateur creosote/coal tar creosote wood preservatives". hse.gov.uk/.
- "Heating Fires in Residential Buildings" (PDF). usfa.dhs.gov/. 2006. Archived from the original (PDF) on 2010-05-27.
- Hodson, E.R. (1906). Rules and Regulations for the Grading of Lumber. Government printing office.
- Howard, Phillip (February 28, 1990). Handbook of Environmental Fate and exposure Data for Organic Chemicals, Volume 2. CRC Press. ISBN 9780873712040. Retrieved October 28, 2016.
- Hunt, George McMonies; Garratt, George Alfred (1967). Wood preservation. McGraw-Hill.
- Ibach, Rebecca E.; Miller, Regis B. (2007). The Encyclopedia of Wood. Skyhorse Publishing Inc.
- Imlay, G. Anderson (1876). "New outlooks in the prophylaxis and treatment of tuberculosis". The Medical Times and Gazette. 2: 514.
- Joerin, A.E. (December 1909). "The seasoning and preservative treatment of wood ties". Popular Mechanics. 48: 767.
- King, John; Felter, Harvey Wickes; Lloyd, John Uri (1905). "Creosote". King's American Dispensatory. 1: 616–617.
- Kinnicutt, Sir Francis P. (1892). "New outlooks in the prophylaxis and treatment of tuberculosis". Boston Medical and Surgical Journal. 126 (21): 513–518. doi:10.1056/nejm189205261262101.
- Lee, Kwang-Guen; Lee, Sung-Eun; Takeoka, Gary R.; Kim, Jeong-Han; Park, Byeoung-Soo (July 2005). "Antioxidant activity and characterization of volatile constituents of beechwood creosote". Journal of the Science of Food and Agriculture. 85 (9): 1580–1586. doi:10.1002/jsfa.2156. Archived from the original on 2012-03-28. Retrieved 2011-07-25.
- Letheby, Henry (1870). On food: its varieties, chemical composition, nutritive value, comparative digestibility, physiological functions and uses, preparation, culinary treatment, preservation, adulteration, etc. Longmans, Green.
- Lovley, DR; Lonergan, DJ (June 1990). "Anaerobic Oxidation of Toluene, Phenol, and p-Cresol by the Dissimilatory Iron-Reducing Organism, GS-15". Applied and Environmental Microbiology. 56 (6): 1858–1864. Bibcode:1990ApEnM..56.1858L. doi:10.1128/AEM.56.6.1858-1864.1990. PMC 184522. PMID 16348226.
- MacLean, J.D. (December 1952). Preservative Treatment of Wood by Pressure Methods (PDF) (Report). United States Department of Agriculture, Forest Service. Handbook No. 40. Retrieved April 7, 2019.
- Martin, Geoffrey (1913). Industrial and manufacturing chemistry: a practical treatise. Vol. 1. Appleton.
- Martin, Stanlisas (1862). "Solidified Creosote". British Journal of Dental Science. 5: 290.
- Melber, Christine; Kielhorn, Janet; Mangelsdorf, Inge (2004). Coal Tar Creosote (PDF) (Report). United Nations Environment Programme, International Labour Organization, and World Health Organization.
- Mueller, J.G.; Chapman, P.J.; Pritchard, P.H. (December 1989). "Action of a Fluoranthene-Utilizing Bacterial Community on Polycyclic Aromatic Hydrocarbon Components of Creosote". Applied and Environmental Microbiology. 55 (12): 3085–90. Bibcode:1989ApEnM..55.3085M. doi:10.1128/AEM.55.12.3085-3090.1989. PMC 203227. PMID 16348069.
- Mushrush, George C.; Speight, J.G. (1995). Petroleum products: instability and incompatibility. CRC Press. ISBN 9781560322979.
- Neff, J.M. (2002). Bioaccumulation in Marine Organisms: Effect of Contamination from Oil Well Produced Water. Elsevier. ISBN 9780080527840. Retrieved October 29, 2016.
- Nelson, Thomas (1907). Nelson's encyclopaedia: everybody's book of reference. Vol. 3. Thomas Nelson.
- Nickels, Benjamin (1890). "Creosote". In Thorpe, Sir Thomas Edward (ed.). A Dictionary of Applied Chemistry. Vol. 1. pp. 614–620.
- Noller, Carl Robert (1965). Chemistry of organic compounds. Saunders.
- Orr, Wilson L.; White, Curt M. (1990). Geochemistry of sulfur in fossil fuels. American Chemical Society. ISBN 9780841218048.
- Parr, Bartholemew (1809). The London Medical Dictionary, including under distinct heads every branch of medecine. Vol. 1. J. Johnson.
- Pease, William A. (1862). "Arsenic, its application and use". British Journal of Dental Science. 5: 417–426.
- Pharmaceutical Society of Great Britain (1898). "Creosotum". Pharmaceutical Journal: A Weekly Record of Pharmacy and Allied Sciences. 61: 468.
- Phelps, CD; Young, LY (February 1999). "Anaerobic biodegradation of BTEX and gasoline in various aquatic sediments". Biodegradation. 10 (1): 15–25. doi:10.1023/a:1008303729431. PMID 10423837. S2CID 23687943.
- Philips, H. Joshua (1891). Engineering chemistry: a practical treatise for the use of analytical chemists, engineers, ironmasters, iron founders, students, and others. C. Lockwood & son.
- Pliny (1855). Pliny's Natural History, Volume 3. H. G. Bohn.
- Pliny (1856). Pliny's Natural History, Volume 5. H. G. Bohn.
- Price, Overton W.; Kellogg, R.S.; Cox, W.T. (1909). Forests of the United States: Their Use. Government printing office.
- Ramanand, K; Suflita, JM (June 1991). "Anaerobic degradation of m-cresol in anoxic aquifer slurries: carboxylation reactions in a sulfate-reducing bacterial enrichment". Applied and Environmental Microbiology. 57 (6): 1689–95. Bibcode:1991ApEnM..57.1689R. doi:10.1128/AEM.57.6.1689-1695.1991. PMC 183453. PMID 1872602.
- Renard, Adolphe (1895). "Pine Tar". Journal of the Chemical Society. 68 (1): 294.
- Richardson, Barry A. (1993). Wood preservation. Taylor & Francis. ISBN 9780419174905.
- Roscoe, Henry Enfield; Schorlemmer, Carl (1888). "Creosote and Creosote oils". A Treatise on Chemistry: The Hydrocarbons and Their Derivatives or Organic Chemistry. 3:4: 32–37.
- Salmon, D.E. (1901). Relationship of bovine tuberculosis to public health. Government printing office.
- Schorlemmer, C. (1885). "The history of creosote, cedriret, and pittacal". Journal of the Society of Chemical Industry. 4: 152–157.
- Seirogan (2011). "A Gift from the Forest". seirogan.co.jp/.
- Shupe, Todd; Lebow, Stan; Ring, Dennis (June 2008). "Causes and Control of Wood Decay, Degradation and Stain" (PDF). LSU Agricultural Center. Retrieved October 28, 2016.
- Shupe, Todd (September 27, 2012). "Marine Wood Borers". LSU Agricultural Center. Archived from the original on 2016-09-05.
- Smith, Stephen (May 31, 2002). "Environmental Issues Related to the Use of Creosote Wood Preservative". AquAeTer. Retrieved October 28, 2016 – via Research Gate.
- Smolenski, WJ; Suflita, JM (April 1987). "Biodegradation of Cresol Isomers in Anoxic Aquifers". Applied and Environmental Microbiology. 53 (4): 710–716. Bibcode:1987ApEnM..53..710S. doi:10.1128/AEM.53.4.710-716.1987. PMC 203742. PMID 3579279.
- Speight, J.G. (1994). The chemistry and technology of coal. CRC Press. ISBN 9780824792008.
- Stimson, Earl (1914). "Report of the committee XVII on wood preservation". Proceedings of the Annual Convention of the American Railway, Bridge and Building Association. 15: 625–633.
- Taylor, C.F. (1902). "Creosote". The Medical World. 20: 207.
- Vitet, Louis (1778). Pharmacopée de Lyon, ou exposition méthodique des médicaments simples et composés. Chez les Freres Perisse.
- Voorhies, Glenn (June 1940). "Oil tar creosote for wood preservation". ir.library.oregonstate.edu.
- Wang F, Yang G, Zhang W, Wu W, Xu J (June 2004). "Oxidation of p-Cresol to p-Hydroxybenzaldehyde with Molecular Oxygen in the Presence of CuMn-Oxide Heterogeneous Catalyst". Advanced Synthesis and Catalysis. 346 (6): 633–638. doi:10.1002/adsc.200303226.
- Weitkamp, Don; Bennett, Jesse (June 2011). Creosote Release from Cut/broken Piles, Asarco Smelter Site (PDF) (Report). Bellevue, WA: Parametrix. Archived from the original (PDF) on 2016-08-12.
- Whittaker, J.T. (1893). "Creosote in Tuberculosis Pulmonum". Transactions of the Association of American Physicians. 8: 77–90.
- Wong, O; Harris, F (July 2005). "Retrospective cohort mortality study and nested case-control study of workers exposed to creosote at 11 wood-treating plants in the United States". J. Occup. Environ. Med. 47 (7): 683–97. doi:10.1097/01.jom.0000165016.71465.7a. PMID 16010195. S2CID 6571472.
- Zazo JA, Casas JA, Mohedano AF, Gilarranz MA, Rodríguez JJ (October 26, 2005). "Chemical Pathway and Kinetics of Phenol Oxidation by Fenton's Reagent". Environmental Science & Technology. 39 (23): 9295–9302. Bibcode:2005EnST...39.9295Z. doi:10.1021/es050452h. PMID 16382955.
External links
Non-timber forest products | |
|---|---|
| Animal products | |
|
|
| Edible plants / roots |
|
| Mushrooms |
|
| |
| |
| Resins |
|
| Sap / Gum / etc. |
|
| Other | |
| Related |
|
| |