Neurodegenerative disease
A neurodegenerative disease is caused by the progressive loss of structure or function of neurons, in the process known as neurodegeneration. Such neuronal damage may ultimately involve cell death. Neurodegenerative diseases include amyotrophic lateral sclerosis, multiple sclerosis, Parkinson's disease, Alzheimer's disease, Huntington's disease, multiple system atrophy, and prion diseases. Neurodegeneration can be found in the brain at many different levels of neuronal circuitry, ranging from molecular to systemic. Because there is no known way to reverse the progressive degeneration of neurons, these diseases are considered to be incurable; however research has shown that the two major contributing factors to neurodegeneration are oxidative stress and inflammation.[2][3][4][5] Biomedical research has revealed many similarities between these diseases at the subcellular level, including atypical protein assemblies (like proteinopathy) and induced cell death.[6][7] These similarities suggest that therapeutic advances against one neurodegenerative disease might ameliorate other diseases as well.
| Neurodegenerative disease | |
|---|---|
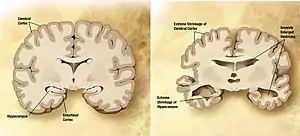 | |
| Normal brain on left contrasted with structural changes shown in brain on right of person with Alzheimer's disease, the most common neurodegenerative disease[1] | |
| Specialty | Neurology, Psychiatry |
Within neurodegenerative diseases, it is estimated that 55 million people worldwide had dementia in 2019, and that by 2050 this figure will increase to 139 million people.[8]
Specific disorders
Alzheimer's disease
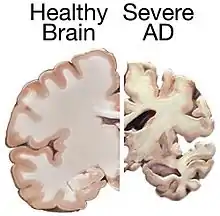
Alzheimer's disease (AD) is a chronic neurodegenerative disease that results in the loss of neurons and synapses in the cerebral cortex and certain subcortical structures, resulting in gross atrophy of the temporal lobe, parietal lobe, and parts of the frontal cortex and cingulate gyrus.[9] It is the most common neurodegenerative disease.[1] Even with billions of dollars being used to find a treatment for Alzheimer's disease, no effective treatments have been found.[10] However, clinical trials have developed certain compounds that could potentially change the future of Alzheimer's disease treatments.[11] Currently, diagnoses of Alzheimer's is subpar, and better methods need to be utilized for various aspects of clinical diagnoses.[12] Alzheimer's has a 20% misdiagnosis rate.[12]
AD pathology is primarily characterized by the presence of amyloid plaques and neurofibrillary tangles. Plaques are made up of small peptides, typically 39–43 amino acids in length, called amyloid beta (also written as A-beta or Aβ). Amyloid beta is a fragment from a larger protein called amyloid precursor protein (APP), a transmembrane protein that penetrates through the neuron's membrane. APP appears to play roles in normal neuron growth, survival and post-injury repair.[13][14] APP is cleaved into smaller fragments by enzymes such as gamma secretase and beta secretase.[15] One of these fragments gives rise to fibrils of amyloid beta which can self-assemble into the dense extracellular amyloid plaques.[16][17]
Parkinson's disease
Parkinson's disease (PD) is the second most common neurodegenerative disorder.[18] It typically manifests as bradykinesia, rigidity, resting tremor and posture instability. The crude prevalence rate of PD has been reported to range from 15 per 100,000 to 12,500 per 100,000, and the incidence of PD from 15 per 100,000 to 328 per 100,000, with the disease being less common in Asian countries.
PD is primarily characterized by death of dopaminergic neurons in the substantia nigra, a region of the midbrain. The cause of this selective cell death is unknown. Notably, alpha-synuclein-ubiquitin complexes and aggregates are observed to accumulate in Lewy bodies within affected neurons. It is thought that defects in protein transport machinery and regulation, such as RAB1, may play a role in this disease mechanism.[19] Impaired axonal transport of alpha-synuclein may also lead to its accumulation in Lewy bodies. Experiments have revealed reduced transport rates of both wild-type and two familial Parkinson's disease-associated mutant alpha-synucleins through axons of cultured neurons.[20] Membrane damage by alpha-synuclein could be another Parkinson's disease mechanism.[21]
The main known risk factor is age. Mutations in genes such as α-synuclein (SNCA), leucine-rich repeat kinase 2 (LRRK2), glucocerebrosidase (GBA), and tau protein (MAPT) can also cause hereditary PD or increase PD risk.[22] While PD is the second most common neurodegenerative disorder, problems with diagnoses still persist.[23] Problems with the sense of smell is a widespread symptom of Parkinson's disease (PD), however, some neurologists question its efficacy.[23] This assessment method is a source of controversy among medical professionals.[23] The gut microbiome might play a role in the diagnosis of PD, and research suggests various ways that could revolutionize the future of PD treatment.[24]
Huntington's disease
Huntington's disease (HD) is a rare autosomal dominant neurodegenerative disorder caused by mutations in the huntingtin gene (HTT). HD is characterized by loss of medium spiny neurons and astrogliosis.[25][26][27] The first brain region to be substantially affected is the striatum, followed by degeneration of the frontal and temporal cortices.[28] The striatum's subthalamic nuclei send control signals to the globus pallidus, which initiates and modulates motion. The weaker signals from subthalamic nuclei thus cause reduced initiation and modulation of movement, resulting in the characteristic movements of the disorder, notably chorea.[29] Huntington's disease presents itself later in life even though the proteins that cause the disease works towards manifestation from their early stages in the humans affected by the proteins.[30] Along with being a neurodegenerative disorder, HD has links to problems with neurodevelopment.[30]
HD is caused by polyglutamine tract expansion in the huntingtin gene, resulting in the mutant huntingtin. Aggregates of mutant huntingtin form as inclusion bodies in neurons, and may be directly toxic. Additionally, they may damage molecular motors and microtubules to interfere with normal axonal transport, leading to impaired transport of important cargoes such as BDNF.[20] Huntington's disease currently has no effective treatments that would modify the disease.[31]
Multiple sclerosis
Multiple sclerosis (MS) is a chronic debilitating demyelinating disease of the central nervous system, caused by an autoimmune attack resulting in the progressive loss of myelin sheath on neuronal axons.[32] The resultant decrease in the speed of signal transduction leads to a loss of functionality that includes both cognitive and motor impairment depending on the location of the lesion.[32] The progression of MS occurs due to episodes of increasing inflammation, which is proposed to be due to the release of antigens such as myelin oligodendrocyte glycoprotein, myelin basic protein, and proteolipid protein, causing an autoimmune response.[33] This sets off a cascade of signaling molecules that result in T cells, B cells, and Macrophages to cross the blood-brain barrier and attack myelin on neuronal axons leading to inflammation.[34] Further release of antigens drives subsequent degeneration causing increased inflammation.[35] Multiple sclerosis presents itself as a spectrum based on the degree of inflammation, a majority of patients experience early relapsing and remitting episodes of neuronal deterioration following a period of recovery. Some of these individuals may transition to a more linear progression of the disease, while about 15% of others begin with a progressive course on the onset of Multiple sclerosis. The inflammatory response contributes to the loss of the grey matter, and as a result current literature devotes itself to combatting the auto-inflammatory aspect of the disease.[34] While there are several proposed causal links between EBV and the HLA-DRB1*15:01 allele to the onset of MS – they may contribute to the degree of autoimmune attack and the resultant inflammation – they do not determine the onset of MS.[34]
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) or Lou Gehrig's disease is a disease in which motor neurons are selectively targeted for degeneration. Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder that negatively impacts the upper motor neurons (UMNs) and lower motor neurons (LMNs).[36] In 1993, missense mutations in the gene encoding the antioxidant enzyme Cu/Zn superoxide dismutase 1 (SOD1) were discovered in a subsets of patients with familial ALS. This discovery led researchers to focus on unlocking the mechanisms for SOD1-mediated diseases. However, the pathogenic mechanism underlying SOD1 mutant toxicity has yet to be resolved. More recently, TDP-43 and FUS protein aggregates have been implicated in some cases of the disease, and a mutation in chromosome 9 (C9orf72) is thought to be the most common known cause of sporadic ALS. It is diagnosed by skeletal muscle weakness that progresses gradually.[36] Early diagnosis of ALS is harder than with other neurodegenerative diseases as there are no highly effective means of determining its early onset.[36] Currently, there is research being done regarding the diagnosis of ALS through upper motor neuron tests.[37] The Penn Upper Motor Neuron Score (PUMNS) consists of 28 criteria with a score range of 0-32.[37] A higher score indicates a higher level of burden present on the upper motor neurons.[37] The PUMNS has proven quite effective in determining the burden that exists on upper motor neurons in affected patients.[37]
Independent research provided in vitro evidence that the primary cellular sites where SOD1 mutations act are located on astrocytes.[38][39] Astrocytes then cause the toxic effects on the motor neurons. The specific mechanism of toxicity still needs to be investigated, but the findings are significant because they implicate cells other than neuron cells in neurodegeneration.[40]
Batten disease
Batten disease is a rare and fatal recessive neurodegenerative disorder that begins in childhood.[41] Batten disease is the common name for a group of lysosomal storage disorders known as neuronal ceroid lipofuscinoses (NCLs) – each caused by a specific gene mutation,[41] of which there are thirteen.[42] Since Batten disease is quite rare, its worldwide prevalence is about 1 in every 100,000 live births.[43] In North America, CLN3 disease (juvenile NCL) typically manifests between the ages of 4 to 7.[44] Batten disease is characterized by motor impairment, epilepsy, dementia, vision loss, and shortened lifespan.[45] A loss of vision is common first sign of Batten disease.[44] Loss of vision is typically preceded by cognitive and behavioral changes, seizures, and loss of the ability to walk.[44] It is common for people to establish cardiac arrhythmias and difficulties eating food as the disease progresses.[44] Batten disease diagnosis depends on a conflation of many criteria: clinical signs and symptoms, evaluations of the eye, electroencephalograms (EEG), and brain magnetic resonance imaging (MRI) results.[43] The diagnosis provided by these results are corroborated by genetic and biochemical testing.[43] No effective treatments were available to prevent the disease from being widespread before the past few years.[43] In recent years, more models have been created to expedite the research process for methods to treat Batten disease.[43]
Creutzfeldt–Jakob disease
Creutzfeldt–Jakob disease (CJD) is a prion disease that is characterized by rapidly progressive dementia.[46] Abnormal proteins called prions aggregate in brain tissue leading to nerve cell death.[47] Prions are misfolded PRNP proteins. They are also infectious. Variant Creutzfeldt–Jakob disease (vCJD) is the infectious form that comes from the meat of a cow that was infected with bovine spongiform encephalopathy, also called mad cow disease.[48]
Risk factor
The greatest risk factor for neurodegenerative diseases is aging. Mitochondrial DNA mutations as well as oxidative stress both contribute to aging.[49] Many of these diseases are late-onset, meaning there is some factor that changes as a person ages for each disease.[6] One constant factor is that in each disease, neurons gradually lose function as the disease progresses with age. It has been proposed that DNA damage accumulation provides the underlying causative link between aging and neurodegenerative disease.[50][51] About 20–40% of healthy people between 60 and 78 years old experience discernable decrements in cognitive performance in several domains including working, spatial, and episodic memory, and processing speed.[52]
Mechanisms
Genetics
Many neurodegenerative diseases are caused by genetic mutations, most of which are located in completely unrelated genes. In many of the different diseases, the mutated gene has a common feature: a repeat of the CAG nucleotide triplet. CAG codes for the amino acid glutamine. A repeat of CAG results in a polyglutamine (polyQ) tract. Diseases associated with such mutations are known as trinucleotide repeat disorders.[53][54]
Polyglutamine repeats typically cause dominant pathogenesis. Extra glutamine residues can acquire toxic properties through a variety of ways, including irregular protein folding and degradation pathways, altered subcellular localization, and abnormal interactions with other cellular proteins.[53] PolyQ studies often use a variety of animal models because there is such a clearly defined trigger – repeat expansion. Extensive research has been done using the models of nematode (C. elegans), and fruit fly (Drosophila), mice, and non-human primates.[54][55]
Nine inherited neurodegenerative diseases are caused by the expansion of the CAG trinucleotide and polyQ tract, including Huntington's disease and the spinocerebellar ataxias.[56]
Epigenetics
The presence of epigenetic modifications for certain genes has been demonstrated in this type of pathology. An example is FKBP5 gene, which progressively increases its expression with age and has been related to Braak staging and increased tau pathology both in vitro and in mouse models of AD.[57]
Protein misfolding
Several neurodegenerative diseases are classified as proteopathies as they are associated with the aggregation of misfolded proteins. Protein toxicity is one of the key mechanisms of many neurodegenrative diseases.[58]
- alpha-synuclein: can aggregate to form insoluble fibrils in pathological conditions characterized by Lewy bodies, such as Parkinson's disease, dementia with Lewy bodies, and multiple system atrophy. Alpha-synuclein is the primary structural component of Lewy body fibrils. In addition, an alpha-synuclein fragment, known as the non-Abeta component (NAC), is found in amyloid plaques in Alzheimer's disease.
- tau: hyperphosphorylated tau protein is the main component of neurofibrillary tangles in Alzheimer's disease; tau fibrils are the main component of Pick bodies found in behavioral variant frontotemporal dementia.
- amyloid beta: the major component of amyloid plaques in Alzheimer's disease.
- prion: main component of prion diseases and transmissible spongiform encephalopathy.
Protein degradation pathways
Parkinson's disease and Huntington's disease are both late-onset and associated with the accumulation of intracellular toxic proteins. Diseases caused by the aggregation of proteins are known as proteopathies, and they are primarily caused by aggregates in the following structures:[6]
- cytosol, e.g. Parkinson's and Huntington's
- nucleus, e.g. Spinocerebellar ataxia type 1 (SCA1)
- endoplasmic reticulum (ER), (as seen with neuroserpin mutations that cause familial encephalopathy with neuroserpin inclusion bodies)
- extracellularly excreted proteins, amyloid-beta in Alzheimer's disease
There are two main avenues eukaryotic cells use to remove troublesome proteins or organelles:
- ubiquitin–proteasome: protein ubiquitin along with enzymes is key for the degradation of many proteins that cause proteopathies including polyQ expansions and alpha-synucleins. Research indicates proteasome enzymes may not be able to correctly cleave these irregular proteins, which could possibly result in a more toxic species. This is the primary route cells use to degrade proteins.[6]
- Decreased proteasome activity is consistent with models in which intracellular protein aggregates form. It is still unknown whether or not these aggregates are a cause or a result of neurodegeneration.[6]
- autophagy–lysosome pathways: a form of programmed cell death (PCD), this becomes the favorable route when a protein is aggregate-prone meaning it is a poor proteasome substrate. This can be split into two forms of autophagy: macroautophagy and chaperone-mediated autophagy (CMA).[6]
- macroautophagy is involved with nutrient recycling of macromolecules under conditions of starvation, certain apoptotic pathways, and if absent, leads to the formation of ubiquinated inclusions. Experiments in mice with neuronally confined macroautophagy-gene knockouts develop intraneuronal aggregates leading to neurodegeneration.[6]
- chaperone-mediated autophagy defects may also lead to neurodegeneration. Research has shown that mutant proteins bind to the CMA-pathway receptors on lysosomal membrane and in doing so block their own degradation as well as the degradation of other substrates.[6]
Membrane damage
Damage to the membranes of organelles by monomeric or oligomeric proteins could also contribute to these diseases. Alpha-synuclein can damage membranes by inducing membrane curvature,[21] and cause extensive tubulation and vesiculation when incubated with artificial phospholipid vesicles.[21] The tubes formed from these lipid vesicles consist of both micellar as well as bilayer tubes. Extensive induction of membrane curvature is deleterious to the cell and would eventually lead to cell death. Apart from tubular structures, alpha-synuclein can also form lipoprotein nanoparticles similar to apolipoproteins.
Mitochondrial dysfunction
The most common form of cell death in neurodegeneration is through the intrinsic mitochondrial apoptotic pathway. This pathway controls the activation of caspase-9 by regulating the release of cytochrome c from the mitochondrial intermembrane space. Reactive oxygen species (ROS) are normal byproducts of mitochondrial respiratory chain activity. ROS concentration is mediated by mitochondrial antioxidants such as manganese superoxide dismutase (SOD2) and glutathione peroxidase. Over production of ROS (oxidative stress) is a central feature of all neurodegenerative disorders. In addition to the generation of ROS, mitochondria are also involved with life-sustaining functions including calcium homeostasis, PCD, mitochondrial fission and fusion, lipid concentration of the mitochondrial membranes, and the mitochondrial permeability transition. Mitochondrial disease leading to neurodegeneration is likely, at least on some level, to involve all of these functions.[59]
There is strong evidence that mitochondrial dysfunction and oxidative stress play a causal role in neurodegenerative disease pathogenesis, including in four of the more well known diseases Alzheimer's, Parkinson's, Huntington's, and amyotrophic lateral sclerosis.[49]
Neurons are particularly vulnerable to oxidative damage due to their strong metabolic activity associated with high transcription levels, high oxygen consumption, and weak antioxidant defense.[60][61]
DNA damage
The brain metabolizes as much as a fifth of consumed oxygen, and reactive oxygen species produced by oxidative metabolism are a major source of DNA damage in the brain. Damage to a cell's DNA is particularly harmful because DNA is the blueprint for protein production and unlike other molecules it cannot simply be replaced by re-synthesis. The vulnerability of post-mitotic neurons to DNA damage (such as oxidative lesions or certain types of DNA strand breaks), coupled with a gradual decline in the activities of repair mechanisms, could lead to accumulation of DNA damage with age and contribute to brain aging and neurodegeneration.[62] DNA single-strand breaks are common and are associated with the neurodegenerative disease ataxia-oculomotor apraxia.[63][61] Increased oxidative DNA damage in the brain is associated with Alzheimer's disease and Parkinson's disease.[63] Defective DNA repair has been linked to neurodegenerative disorders such as Alzheimer's disease, amyotrophic lateral sclerosis, ataxia telangiectasia, Cockayne syndrome, Parkinson's disease and xeroderma pigmentosum.[63][62]
Axonal transport
Axonal swelling, and axonal spheroids have been observed in many different neurodegenerative diseases. This suggests that defective axons are not only present in diseased neurons, but also that they may cause certain pathological insult due to accumulation of organelles. Axonal transport can be disrupted by a variety of mechanisms including damage to: kinesin and cytoplasmic dynein, microtubules, cargoes, and mitochondria.[20] When axonal transport is severely disrupted a degenerative pathway known as Wallerian-like degeneration is often triggered.[64]
Programmed cell death
Programmed cell death (PCD) is death of a cell in any form, mediated by an intracellular program.[65] This process can be activated in neurodegenerative diseases including Parkinson's disease, amytrophic lateral sclerosis, Alzheimer's disease and Huntington's disease.[66] PCD observed in neurodegenerative diseases may be directly pathogenic; alternatively, PCD may occur in response to other injury or disease processes.[7]
Apoptosis (type I)
Apoptosis is a form of programmed cell death in multicellular organisms. It is one of the main types of programmed cell death (PCD) and involves a series of biochemical events leading to a characteristic cell morphology and death.
- Extrinsic apoptotic pathways: Occur when factors outside the cell activate cell surface death receptors (e.g., Fas) that result in the activation of caspases-8 or -10.[7]
- Intrinsic apoptotic pathways: Result from mitochondrial release of cytochrome c or endoplasmic reticulum malfunctions, each leading to the activation of caspase-9. The nucleus and Golgi apparatus are other organelles that have damage sensors, which can lead the cells down apoptotic pathways.[7][67]
Caspases (cysteine-aspartic acid proteases) cleave at very specific amino acid residues. There are two types of caspases: initiators and effectors. Initiator caspases cleave inactive forms of effector caspases. This activates the effectors that in turn cleave other proteins resulting in apoptotic initiation.[7]
Autophagic (type II)
Autophagy is a form of intracellular phagocytosis in which a cell actively consumes damaged organelles or misfolded proteins by encapsulating them into an autophagosome, which fuses with a lysosome to destroy the contents of the autophagosome. Because many neurodegenerative diseases show unusual protein aggregates, it is hypothesized that defects in autophagy could be a common mechanism of neurodegeneration.[7]
Cytoplasmic (type III)
PCD can also occur via non-apoptotic processes, also known as Type III or cytoplasmic cell death. For example, type III PCD might be caused by trophotoxicity, or hyperactivation of trophic factor receptors. Cytotoxins that induce PCD can cause necrosis at low concentrations, or aponecrosis (combination of apoptosis and necrosis) at higher concentrations. It is still unclear exactly what combination of apoptosis, non-apoptosis, and necrosis causes different kinds of aponecrosis.[7]
Transglutaminase
Transglutaminases are human enzymes ubiquitously present in the human body and in the brain in particular.[68]
The main function of transglutaminases is bind proteins and peptides intra- and intermolecularly, by a type of covalent bonds termed isopeptide bonds, in a reaction termed transamidation or crosslinking.[68]
Transglutaminase binding of these proteins and peptides make them clump together. The resulting structures are turned extremely resistant to chemical and mechanical disruption.[68]
Most relevant human neurodegenerative diseases share the property of having abnormal structures made up of proteins and peptides.[68]
Each of these neurodegenerative diseases have one (or several) specific main protein or peptide. In Alzheimer's disease, these are amyloid-beta and tau. In Parkinson's disease, it is alpha-synuclein. In Huntington's disease, it is huntingtin.[68]
Transglutaminase substrates: Amyloid-beta, tau, alpha-synuclein and huntingtin have been proved to be substrates of transglutaminases in vitro or in vivo, that is, they can be bonded by trasglutaminases by covalent bonds to each other and potentially to any other transglutaminase substrate in the brain.[68]
Transglutaminase augmented expression: It has been proved that in these neurodegenerative diseases (Alzheimer's disease, Parkinson's disease, and Huntington's disease) the expression of the transglutaminase enzyme is increased.[68]
Presence of isopeptide bonds in these structures: The presence of isopeptide bonds (the result of the transglutaminase reaction) have been detected in the abnormal structures that are characteristic of these neurodegenerative diseases.[68]
Co-localization: Co-localization of transglutaminase mediated isopeptide bonds with these abnormal structures has been detected in the autopsy of brains of patients with these diseases.[68]
Management
The process of neurodegeneration is not well understood, so the diseases that stem from it have, as yet, no cures.
Animal models in research
In the search for effective treatments (as opposed to palliative care), investigators employ animal models of disease to test potential therapeutic agents. Model organisms provide an inexpensive and relatively quick means to perform two main functions: target identification and target validation. Together, these help show the value of any specific therapeutic strategies and drugs when attempting to ameliorate disease severity. An example is the drug Dimebon by Medivation, Inc. In 2009 this drug was in phase III clinical trials for use in Alzheimer's disease, and also phase II clinical trials for use in Huntington's disease.[54] In March 2010, the results of a clinical trial phase III were released; the investigational Alzheimer's disease drug Dimebon failed in the pivotal CONNECTION trial of patients with mild-to-moderate disease.[69] With CONCERT, the remaining Pfizer and Medivation Phase III trial for Dimebon (latrepirdine) in Alzheimer's disease failed in 2012, effectively ending the development in this indication.[70]
In another experiment using a rat model of Alzheimer's disease, it was demonstrated that systemic administration of hypothalamic proline-rich peptide (PRP)-1 offers neuroprotective effects and can prevent neurodegeneration in hippocampus amyloid-beta 25–35. This suggests that there could be therapeutic value to PRP-1.[71]
Other avenues of investigation
Protein degradation offers therapeutic options both in preventing the synthesis and degradation of irregular proteins. There is also interest in upregulating autophagy to help clear protein aggregates implicated in neurodegeneration. Both of these options involve very complex pathways that we are only beginning to understand.[6]
The goal of immunotherapy is to enhance aspects of the immune system. Both active and passive vaccinations have been proposed for Alzheimer's disease and other conditions; however, more research must be done to prove safety and efficacy in humans.[72]
A current therapeutic target for the treatment of Alzheimer's disease is the protease β-secretase[73], which is involved in the amyloidogenic processing pathway that leads to the pathological accumulation of proteins in the brain. When the gene that encodes for amyloid precursor protein (APP) is spliced by α-secretase[74] rather than β-secretase, the toxic protein β amyloid is not produced. Targeted inhibition[75] of β-secretase can potentially prevent the neuronal death that is responsible for the symptoms of Alzheimer's disease.
See also
- Amyloid
- JUNQ and IPOD
- Neurodegeneration with brain iron accumulation
- Prevention of dementia
References
- Erkkinen, Michael G.; Kim, Mee-Ohk; Geschwind, Michael D (April 2018). "Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases". Cold Spring Harbor Perspectives in Biology. 10 (4): 20. doi:10.1101/cshperspect.a033118. PMC 5880171. PMID 28716886.
- Pereira, TMC; Côco, LZ; Ton, AMM; Meyrelles, SS; Campos-Toimil, M; Campagnaro, BP; Vasquez, EC (20 November 2021). "The Emerging Scenario of the Gut-Brain Axis: The Therapeutic Actions of the New Actor Kefir against Neurodegenerative Diseases". Antioxidants (Basel, Switzerland). 10 (11): 1845. doi:10.3390/antiox10111845. PMC 8614795. PMID 34829716.
- Stephenson, J; Nutma, E; van der Valk, P; Amor, S (June 2018). "Inflammation in CNS neurodegenerative diseases". Immunology. 154 (2): 204–219. doi:10.1111/imm.12922. PMC 5980185. PMID 29513402.
- Singh, A; Kukreti, R; Saso, L; Kukreti, S (22 April 2019). "Oxidative Stress: A Key Modulator in Neurodegenerative Diseases". Molecules (Basel, Switzerland). 24 (8): 1583. doi:10.3390/molecules24081583. PMC 6514564. PMID 31013638.
- "What is Neurodegenerative Disease?". JPND Research. JPND Research. 17 July 2014. Retrieved February 7, 2015.
- Rubinsztein DC (October 2006). "The roles of intracellular protein-degradation pathways in neurodegeneration". Nature. 443 (7113): 780–6. Bibcode:2006Natur.443..780R. doi:10.1038/nature05291. PMID 17051204. S2CID 4411895.
- Bredesen DE, Rao RV, Mehlen P (October 2006). "Cell death in the nervous system". Nature. 443 (7113): 796–802. Bibcode:2006Natur.443..796B. doi:10.1038/nature05293. PMC 3970704. PMID 17051206.
- Global status report on the public health response to dementia (PDF). Geneva: World Health Organization. 2021. ISBN 978-92-4-003324-5. Retrieved 14 October 2022.
- Wenk GL (2003). "Neuropathologic changes in Alzheimer's disease". The Journal of Clinical Psychiatry. 64 Suppl 9: 7–10. PMID 12934968.
- Lock MM (27 October 2013). The Alzheimer conundrum : entanglements of dementia and aging. Princeton. ISBN 978-1-4008-4846-1. OCLC 859536969.
- Delrieu J, Ousset PJ, Caillaud C, Vellas B (January 2012). "'Clinical trials in Alzheimer's disease': immunotherapy approaches". Journal of Neurochemistry. 120 Suppl 1: 186–93. doi:10.1111/j.1471-4159.2011.07458.x. PMID 21883222.
- Archer MC, Hall PH, Morgan JC (2017). "[P2–430]: Accuracy of Clinical Diagnosis of Alzheimer's Disease in Alzheimer's Disease Centers (ADCS)". Alzheimer's & Dementia. 13 (7S_Part_16): P800–P801. doi:10.1016/j.jalz.2017.06.1086. S2CID 54359937.
- Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J (July 2006). "Synapse formation and function is modulated by the amyloid precursor protein". The Journal of Neuroscience. 26 (27): 7212–21. doi:10.1523/JNEUROSCI.1450-06.2006. PMC 6673945. PMID 16822978.
- Turner PR, O'Connor K, Tate WP, Abraham WC (May 2003). "Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory". Progress in Neurobiology. 70 (1): 1–32. doi:10.1016/S0301-0082(03)00089-3. PMID 12927332. S2CID 25376584.
- Hooper NM (April 2005). "Roles of proteolysis and lipid rafts in the processing of the amyloid precursor protein and prion protein". Biochemical Society Transactions. 33 (Pt 2): 335–8. doi:10.1042/BST0330335. PMID 15787600.
- Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J (June 2004). "The importance of neuritic plaques and tangles to the development and evolution of AD". Neurology. 62 (11): 1984–9. doi:10.1212/01.WNL.0000129697.01779.0A. PMID 15184601. S2CID 25017332.
- Ohnishi S, Takano K (March 2004). "Amyloid fibrils from the viewpoint of protein folding". Cellular and Molecular Life Sciences. 61 (5): 511–524. doi:10.1007/s00018-003-3264-8. PMID 15004691. S2CID 25739126.
- Elbaz A, Carcaillon L, Kab S, Moisan F (January 2016). "Epidemiology of Parkinson's disease". Revue Neurologique. 172 (1): 14–26. doi:10.1016/j.neurol.2015.09.012. PMID 26718594.
- "Parkinson's Disease Mechanism Discovered," HHMI Research News June 22, 2006.
- De Vos KJ, Grierson AJ, Ackerley S, Miller CC (2008). "Role of axonal transport in neurodegenerative diseases". Annual Review of Neuroscience. 31: 151–73. doi:10.1146/annurev.neuro.31.061307.090711. PMID 18558852.
- Varkey J, Isas JM, Mizuno N, Jensen MB, Bhatia VK, Jao CC, et al. (October 2010). "Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins". The Journal of Biological Chemistry. 285 (42): 32486–93. doi:10.1074/jbc.M110.139576. PMC 2952250. PMID 20693280.
- Davis AA, Andruska KM, Benitez BA, Racette BA, Perlmutter JS, Cruchaga C (January 2016). "Variants in GBA, SNCA, and MAPT influence Parkinson disease risk, age at onset, and progression". Neurobiology of Aging. 37: 209.e1–209.e7. doi:10.1016/j.neurobiolaging.2015.09.014. PMC 4688052. PMID 26601739.
- Schmidt, Nele; Paschen, Laura; Witt, Karsten (2020-11-16). Mirabella, Giovanni (ed.). "Invalid Self-Assessment of Olfactory Functioning in Parkinson's Disease Patients May Mislead the Neurologist". Parkinson's Disease. 2020: 1–5. doi:10.1155/2020/7548394. ISSN 2042-0080. PMC 7683170. PMID 33274040.
- Hill-Burns, Erin M.; Debelius, Justine W.; Morton, James T.; Wissemann, William T.; Lewis, Matthew R.; Wallen, Zachary D.; Peddada, Shyamal D.; Factor, Stewart A.; Molho, Eric; Zabetian, Cyrus P.; Knight, Rob (May 2017). "Parkinson's disease and Parkinson's disease medications have distinct signatures of the gut microbiome: PD, Medications, and Gut Microbiome". Movement Disorders. 32 (5): 739–749. doi:10.1002/mds.26942. PMC 5469442. PMID 28195358.
- Purves D, Augustine GA, Fitzpatrick D, Hall W, LaMantia AS, McNamara JO, Williams SM (2001). "Modulation of Movement by the Basal Ganglia - Circuits within the Basal Ganglia System". In Dale Purves (ed.). Neuroscience (2nd ed.). Sunderland, MA: Sinauer Associates. ISBN 978-0-87893-742-4.
- Estrada Sánchez AM, Mejía-Toiber J, Massieu L (April 2008). "Excitotoxic neuronal death and the pathogenesis of Huntington's disease". Archives of Medical Research. 39 (3): 265–76. doi:10.1016/j.arcmed.2007.11.011. PMID 18279698.
- Lobsiger CS, Cleveland DW (November 2007). "Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease". Nature Neuroscience. 10 (11): 1355–60. doi:10.1038/nn1988. PMC 3110080. PMID 17965655.
- Purves D, Augustine GA, Fitzpatrick D, Hall W, LaMantia AS, McNamara JO, Williams SM (2001). "Modulation of Movement by the Basal Ganglia - Box A. Huntington's Disease". In Dale Purves (ed.). Neuroscience (2nd ed.). Sunderland, MA: Sinauer Associates. ISBN 978-0-87893-742-4.
- Crossman AR (May 2000). "Functional anatomy of movement disorders". Journal of Anatomy. 196 ( Pt 4) (4): 519–25. doi:10.1046/j.1469-7580.2000.19640519.x. PMC 1468094. PMID 10923984.
- Barnat, Monia; Capizzi, Mariacristina; Aparicio, Esther; Boluda, Susana; Wennagel, Doris; Kacher, Radhia; Kassem, Rayane; Lenoir, Sophie; Agasse, Fabienne; Braz, Barbara Y.; Liu, Jeh-Ping (2020-08-14). "Huntington's disease alters human neurodevelopment". Science. 369 (6505): 787–793. Bibcode:2020Sci...369..787B. doi:10.1126/science.aax3338. ISSN 0036-8075. PMC 7859879. PMID 32675289.
- Labbadia J, Morimoto RI (August 2013). "Huntington's disease: underlying molecular mechanisms and emerging concepts". Trends in Biochemical Sciences. 38 (8): 378–85. doi:10.1016/j.tibs.2013.05.003. PMC 3955166. PMID 23768628.
- "Multiple Sclerosis: Hope Through Research | National Institute of Neurological Disorders and Stroke". www.ninds.nih.gov. Retrieved 2020-11-30.
- Kaufman, David Myland; Milstein, Mark J. (2013-01-01), Kaufman, David Myland; Milstein, Mark J. (eds.), "Chapter 15 - Multiple Sclerosis", Kaufman's Clinical Neurology for Psychiatrists (Seventh Edition), Philadelphia: W.B. Saunders, pp. 329–349, ISBN 978-0-7234-3748-2, retrieved 2020-12-07
- Stys, Peter K.; Tsutsui, Shigeki (2019-12-13). "Recent advances in understanding multiple sclerosis". F1000Research. 8: 2100. doi:10.12688/f1000research.20906.1. ISSN 2046-1402. PMC 6915812. PMID 31885862.
- Teunissen, Charlotte (2008-07-08). "Faculty Opinions recommendation of Remyelination protects axons from demyelination-associated axon degeneration". Brain: A Journal of Neurology. 131 (Pt 6). doi:10.3410/f.1115522.571562. Retrieved 2020-12-07.
- Min YG, Choi SJ, Hong YH, Kim SM, Shin JY, Sung JJ (September 2020). "Dissociated leg muscle atrophy in amyotrophic lateral sclerosis/motor neuron disease: the 'split-leg' sign". Scientific Reports. 10 (1): 15661. Bibcode:2020NatSR..1015661M. doi:10.1038/s41598-020-72887-7. PMC 7518279. PMID 32973334.
- Quinn C, Edmundson C, Dahodwala N, Elman L (April 2020). "Reliable and efficient scale to assess upper motor neuron disease burden in amyotrophic lateral sclerosis". Muscle & Nerve. 61 (4): 508–511. doi:10.1002/mus.26764. PMID 31743477. S2CID 208186566.
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S (May 2007). "Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons". Nature Neuroscience. 10 (5): 615–22. doi:10.1038/nn1876. PMC 3799799. PMID 17435755.
- Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K (May 2007). "Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model". Nature Neuroscience. 10 (5): 608–14. doi:10.1038/nn1885. PMC 3139463. PMID 17435754.
- Julien JP (May 2007). "ALS: astrocytes move in as deadly neighbors". Nature Neuroscience. 10 (5): 535–7. doi:10.1038/nn0507-535. PMID 17453052. S2CID 2987257.
- "Batten Disease Fact Sheet | National Institute of Neurological Disorders and Stroke". www.ninds.nih.gov. Retrieved 30 November 2020.
- Johnson TB, Cain JT, White KA, Ramirez-Montealegre D, Pearce DA, Weimer JM (March 2019). "Therapeutic landscape for Batten disease: current treatments and future prospects". Nature Reviews. Neurology. 15 (3): 161–178. doi:10.1038/s41582-019-0138-8. PMC 6681450. PMID 30783219.
- Johnson, Tyler B.; Cain, Jacob T.; White, Katherine A.; Ramirez-Montealegre, Denia; Pearce, David A.; Weimer, Jill M. (March 2019). "Therapeutic landscape for Batten disease: current treatments and future prospects". Nature Reviews Neurology. 15 (3): 161–178. doi:10.1038/s41582-019-0138-8. ISSN 1759-4758. PMC 6681450. PMID 30783219.
- Masten MC, Williams JD, Vermilion J, Adams HR, Vierhile A, Collins A, et al. (June 2020). "The CLN3 Disease Staging System: A new tool for clinical research in Batten disease". Neurology. 94 (23): e2436–e2440. doi:10.1212/WNL.0000000000009454. PMC 7455368. PMID 32300063.
- Hartnett L (2019-09-30). "Batten disease". Learning Disability Practice. 22 (5): 22. doi:10.7748/ldp.22.5.22.s16. S2CID 241832253.
- "Creutzfeldt-Jakob Disease Fact Sheet | National Institute of Neurological Disorders and Stroke". www.ninds.nih.gov. National Institute of Health. Retrieved 31 March 2022.
- "Creutzfeldt-Jakob disease - Symptoms and causes". Mayo Clinic. Retrieved 31 March 2022.
- Research, Center for Biologics Evaluation and (12 April 2019). "Variant Creutzfeldt-Jakob Disease (vCJD) and Factor VIII (pdFVIII) Questions and Answers". FDA. Retrieved 31 March 2022.
- Lin MT, Beal MF (October 2006). "Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases". Nature. 443 (7113): 787–95. Bibcode:2006Natur.443..787L. doi:10.1038/nature05292. PMID 17051205. S2CID 4421515.
- Bernstein C, Bernstein H (1991). Aging, Sex, and DNA Repair. San Diego: Academic Press. pp. 121–139. ISBN 978-0120928606.
- Maynard S, Fang EF, Scheibye-Knudsen M, Croteau DL, Bohr VA (September 2015). "DNA Damage, DNA Repair, Aging, and Neurodegeneration". Cold Spring Harbor Perspectives in Medicine. 5 (10): a025130. doi:10.1101/cshperspect.a025130. PMC 4588127. PMID 26385091.
- Camandola S, Mattson MP (June 2017). "Brain metabolism in health, aging, and neurodegeneration". The EMBO Journal. 36 (11): 1474–1492. doi:10.15252/embj.201695810. PMC 5452017. PMID 28438892.
- Thompson LM (April 2008). "Neurodegeneration: a question of balance". Nature. 452 (7188): 707–8. Bibcode:2008Natur.452..707T. doi:10.1038/452707a. PMID 18401401. S2CID 205037169.
- Marsh JL, Lukacsovich T, Thompson LM (March 2009). "Animal models of polyglutamine diseases and therapeutic approaches". The Journal of Biological Chemistry. 284 (12): 7431–5. doi:10.1074/jbc.R800065200. PMC 2658038. PMID 18957429.
- Orr HT (March 2009). "Unstable nucleotide repeat minireview series: a molecular biography of unstable repeat disorders". The Journal of Biological Chemistry. 284 (12): 7405. doi:10.1074/jbc.R800067200. PMC 2658033. PMID 18957428.
- Zoghbi HY, Orr HT (March 2009). "Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, spinocerebellar ataxia type 1". The Journal of Biological Chemistry. 284 (12): 7425–9. doi:10.1074/jbc.R800041200. PMC 2658037. PMID 18957430.
- Nabais, Marta F.; Laws, Simon M.; Lin, Tian; Vallerga, Costanza L.; Armstrong, Nicola J.; Blair, Ian P.; Kwok, John B.; Mather, Karen A.; Mellick, George D.; Sachdev, Perminder S.; Wallace, Leanne (2021-03-26). "Meta-analysis of genome-wide DNA methylation identifies shared associations across neurodegenerative disorders". Genome Biology. 22 (1): 90. doi:10.1186/s13059-021-02275-5. ISSN 1474-760X. PMC 8004462. PMID 33771206.
- Chung, CG; Lee, H; Lee, SB (September 2018). "Mechanisms of protein toxicity in neurodegenerative diseases". Cellular and Molecular Life Sciences. 75 (17): 3159–3180. doi:10.1007/s00018-018-2854-4. PMC 6063327. PMID 29947927.
- DiMauro S, Schon EA (2008). "Mitochondrial disorders in the nervous system". Annual Review of Neuroscience. 31: 91–123. doi:10.1146/annurev.neuro.30.051606.094302. PMID 18333761.
- Liu Z, Zhou T, Ziegler AC, Dimitrion P, Zuo L (2017). "Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications". Oxidative Medicine and Cellular Longevity. 2017: 2525967. doi:10.1155/2017/2525967. PMC 5529664. PMID 28785371.
- Wang H, Dharmalingam P, Vasquez V, Mitra J, Boldogh I, Rao KS, et al. (January 2017). "Chronic oxidative damage together with genome repair deficiency in the neurons is a double whammy for neurodegeneration: Is damage response signaling a potential therapeutic target?". Mechanisms of Ageing and Development. 161 (Pt A): 163–176. doi:10.1016/j.mad.2016.09.005. PMC 5316312. PMID 27663141.
- Madabhushi R, Pan L, Tsai LH (July 2014). "DNA damage and its links to neurodegeneration". Neuron. 83 (2): 266–282. doi:10.1016/j.neuron.2014.06.034. PMC 5564444. PMID 25033177.
- Jeppesen DK, Bohr VA, Stevnsner T (July 2011). "DNA repair deficiency in neurodegeneration". Progress in Neurobiology. 94 (2): 166–200. doi:10.1016/j.pneurobio.2011.04.013. PMC 3123739. PMID 21550379.
- Coleman MP & Freeman MF 'Wallerian degeneration, WldS and Nmnat' Annual Review of Neuroscience 2010, 33: 245-67
- Engelberg-Kulka H, Amitai S, Kolodkin-Gal I, Hazan R (October 2006). "Bacterial programmed cell death and multicellular behavior in bacteria". PLOS Genetics. 2 (10): e135. doi:10.1371/journal.pgen.0020135. PMC 1626106. PMID 17069462.
- Vila M, Przedborski S (May 2003). "Targeting programmed cell death in neurodegenerative diseases". Nature Reviews. Neuroscience. 4 (5): 365–75. doi:10.1038/nrn1100. PMID 12728264. S2CID 33018251.
- Green DR, Kroemer G (October 2005). "Pharmacological manipulation of cell death: clinical applications in sight?". The Journal of Clinical Investigation. 115 (10): 2610–7. doi:10.1172/JCI26321. PMC 1236695. PMID 16200193.
- Caccamo D, Currò M, Condello S, Ferlazzo N, Ientile R (February 2010). "Critical role of transglutaminase and other stress proteins during neurodegenerative processes". Amino Acids. 38 (2): 653–8. doi:10.1007/s00726-009-0428-3. PMID 19960212. S2CID 19739739.
- Dimebon Disappoints in Phase 3 Trial
- Sweetlove M: Phase III CONCERT Trial of Latrepirdine. Negative results. Pharm Med 2012;26(2):113-115
- Galoyan AA, Sarkissian JS, Chavushyan VA, Meliksetyan IB, Avagyan ZE, Poghosyan MV, et al. (September 2008). "Neuroprotection by hypothalamic peptide proline-rich peptide-1 in Abeta25-35 model of Alzheimer's disease". Alzheimer's & Dementia. 4 (5): 332–44. doi:10.1016/j.jalz.2007.10.019. PMID 18790460. S2CID 39817779.
- Brody DL, Holtzman DM (2008). "Active and passive immunotherapy for neurodegenerative disorders". Annual Review of Neuroscience. 31: 175–93. doi:10.1146/annurev.neuro.31.060407.125529. PMC 2561172. PMID 18352830.
- Pastorino L, Ikin AF, Lamprianou S, Vacaresse N, Revelli JP, Platt K, et al. (April 2004). "BACE (beta-secretase) modulates the processing of APLP2 in vivo". Molecular and Cellular Neurosciences. 25 (4): 642–9. doi:10.1016/j.mcn.2003.12.013. PMID 15080893. S2CID 54334969.
- Esch FS, Keim PS, Beattie EC, Blacher RW, Culwell AR, Oltersdorf T, et al. (June 1990). "Cleavage of amyloid beta peptide during constitutive processing of its precursor". Science. 248 (4959): 1122–4. Bibcode:1990Sci...248.1122E. doi:10.1126/science.2111583. PMID 2111583.
- Schenk D, Basi GS, Pangalos MN (September 2012). "Treatment strategies targeting amyloid β-protein". Cold Spring Harbor Perspectives in Medicine. 2 (9): a006387. doi:10.1101/cshperspect.a006387. PMC 3426815. PMID 22951439.
| ||
| Basic science |
|  |
| Clinical neuroscience |
| |
| Cognitive neuroscience |
| |
| Interdisciplinary fields |
| |
| Concepts |
| |
| ||