Oncogénesis
Los términos oncogénesis o carcinogénesis hacen referencia literal al proceso por el cual se produce el cáncer. Es el proceso por el cual una célula normal se convierte en una célula cancerosa. Se caracteriza por la progresión de varios cambios celulares a nivel del material genético que finalmente desemboca en la reprogramación de la célula provocando que se reproduzca de manera descontrolada, formando una masa maligna.[1]
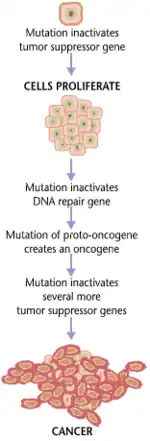
La división celular es un proceso fisiológico que ocurre en casi todos los tejidos y bajo diversas circunstancias. Bajo circunstancias normales, el balance entre la proliferación y la muerte celular programada, usualmente por medio del mecanismo de apoptosis se mantiene estrechamente regulado asegurando de esta forma la integridad de órganos y tejidos. Las mutaciones en el ADN que pueden conducir a una transformación cancerosa interfieren con este proceso de control interfiriendo con el programa que lo controla.
El proceso de oncogénesis es causado por estas mutaciones en el material genético de las células normales, que alteran el balance normal entre proliferación y muerte celular. Como resultado se produce una división celular descontrolada y un proceso evolutivo de estas células por medio de selección natural dentro del organismo. La reproducción rápida y descontrolada de células pueden producir tumores benignos y algunos tipos de estos tumores pueden convertirse en malignos que es lo que se conoce como cáncer. Los tumores benignos no se esparcen a lugares lejanos del organismo ni invaden otros tejidos, y por lo general no representan una amenaza para la vida a menos que compriman estructuras vitales o tengan alguna actividad fisiológica (por ejemplo, que sean capaces de producir alguna hormona). Los tumores malignos son capaces de invadir otros órganos, esparcirse a lugares distantes (proceso conocido como metástasis) y convertirse en una amenaza para la vida.
Generalidades
El cáncer es, fundamentalmente, una enfermedad de regulación del crecimiento de tejidos. Para que una célula normal se transforme en una célula cancerosa, se deben alterar los genes que controlan el crecimiento y la diferenciación celular.[2] Estos cambios genéticos pueden ocurrir en múltiples niveles, desde la pérdida o ganancia de cromosomas completos, a una mutación puntual que afecta un único nucleótido en el ADN. Existen, sin embargo, dos grandes categorías de genes que son afectados por estos cambios. Los oncogenes pueden ser por ejemplo genes normales que son expresados en niveles anormalmente altos, o genes alterados que presentan nuevas propiedades. En cada caso, la expresión de estos genes promueve un fenotipo maligno de células cancerosas. Los genes supresores de tumores son genes que inhiben la supervivencia, la división celular, u otras propiedades de las células cancerosas. Los genes supresores tumorales a menudo son desactivados por cambios genéticos que favorecen la aparición de transformaciones malignas. Típicamente, se requieren cambios en muchos genes para que una célula normal se transforme en una célula cancerosa.[3]
Se requiere más de una mutación para que se produzca la oncogénesis. De hecho, se requiere toda una serie de mutaciones en una cierta clase de genes para que una célula normal se transforme en una célula cancerosa.[4] Solo las mutaciones en aquellos genes que juegan un determinado papel vital en la división celular, en el control de la apoptosis y en la reparación del ADN pueden causar que la célula pierda el control de su propia proliferación.
Existen varios esquemas de clasificación para aquellos cambios genómicos que pueden contribuir a la generación de células cancerosas. La mayor parte de estos cambios son mutaciones, o cambios en la secuencia de nucleótidos del ADN. La aneuploidía, es decir, la presencia de un número anormal de cromosomas, es un cambio genómico que no es una mutación, y puede involucrar tanto la ganancia como la pérdida de uno o más cromosomas debido a errores en la mitosis.
Las mutaciones a gran escala involucran la pérdida o la ganancia de un fragmento de cromosoma. La amplificación génica se produce cuando una célula adquiere múltiples copias (a menudo 20 o más) de una pequeña región cromosómica, que usualmente contiene uno o más oncogenes y material genético adyacente. Las translocaciones cromosómicas se producen cuando dos regiones de cromosomas separados se fusionan de manera anormal, a menudo en una región característica. Un ejemplo muy bien conocido de esto es el cromosoma Filadelfia, o la translocación de los cromosomas 9 y 22, que ocurre en la leucemia mieloide crónica, y que resulta en la producción de la proteína de fusión BCR-Abl, una tirosina quinasa oncogénica.
Los oncovirus, que son un tipo de retrovirus que contienen oncogenes se han categorizado como oncogénicos debido a que son capaces de gatillar el crecimiento de tejidos tumorales en su huésped. Este proceso es a veces referido como transformación viral.
Las mutaciones a pequeña escala incluyen a las mutaciones puntuales, deleciones e inserciones, las cuales pueden ocurrir en el promotor de un determinado gen y de ese modo afectar su expresión, o puede ocurrir en la región codificante del gen y alterar la función o estabilidad de su producto proteico. La disrupción de un único gen puede ocurrir también como resultado de la inserción de material genético viral de un virus de ADN o de un retrovirus, y este evento puede provocar la expresión de oncogenes virales en la célula afectada y en sus descendientes.
Causas
Es, de momento, imposible determinar la causa inicial de la mayor parte de los cánceres. En un par de casos, solo existe una única causa; por ejemplo, el virus HHV-8 causa todos los tipos de sarcoma de Kaposi. Sin embargo, con la ayuda de técnicas de epidemiología del cáncer e información, es posible obtener una causa estimada o mayormente probable en muchas situaciones. Por ejemplo, el cáncer de pulmón puede tener numerosas causas, entre las que se incluyen la exposición al gas radón y el uso de tabaco. Las personas que son fumadoras de tabaco tienen una probabilidad de desarrollar cáncer de pulmón que es 14 veces mayor que la de una persona que nunca fumó tabaco, de modo que la probabilidad de que el cáncer de pulmón de una persona fumadora sea provocado por el tabaco es de alrededor del 93 %; esto significa que el cáncer de pulmón en una persona fumadora tiene una posibilidad de solo el 7 % de ser provocado por alguna otra causa no relacionada con el tabaco, tal como, por ejemplo; el gas radón.[5] Este tipo de correlaciones estadísticas han hecho posible que los investigadores hicieran inferencias acerca de que ciertas sustancias o comportamientos predisponen al desarrollo de cáncer, o son carcinogénicos.
Utilizando técnicas de biología molecular es posible caracterizar las mutaciones o aberraciones cromosomales que poseen las células de un determinado tumor, y se ha hecho un rápido progreso en el área de la predicción (prognosis) del progreso de determinados tipos de tumores con base en el espectro de mutaciones que lo caracteriza. Por ejemplo, más de la mitad de todos los cánceres poseen un gen p53 defectuoso. Este tipo de mutación se asocia a un pobre pronóstico, debido a que es muy poco probable que estas células tumorales entren en un proceso de apoptosis al ser dañadas por la terapia. Las mutaciones en el gen de la telomerasa por ejemplo, aumentan la potencialidad reproductiva de las células tumorales, extendiendo el número de veces que una célula se puede dividir. Otro tipo de mutaciones capacitan al tumor a desarrollar nuevos vasos sanguíneos, para proveerse de más nutrientes, o lo capacitan para metastatizar, esparciéndose a diferentes partes del organismo.
Teorías no convencionales
Hay varias teorías acerca del proceso de oncogénesis y del tratamiento del cáncer que caen fuera de la corriente principal de opiniones de la comunidad científica, ya sea debido a la falta de fundamento científico, lógica, o evidencia de base. Estas teorías pueden ser utilizadas para justificar varios tipos de tratamientos alternativos para el cáncer. Estas teorías deben ser distinguidas de aquellas teorías sobre la oncogénesis que sí poseen una base lógica dentro de lo que se considera la biología del cáncer clásica, y dentro de la cual si pueden establecerse hipótesis contrastables con la evidencia.
Existen, sin embargo, varias teorías no clásicas en cuanto a la oncogénesis que si se basan en evidencia científica, y que cada vez están cobrando mayor atención. Por ejemplo, algunos investigadores sostienen que el cáncer puede ser causado por algunas alteraciones epigenéticas, es decir por alteraciones heredables y reversibles que no se encuentran explicadas por cambios en la secuencia de ADN de las células,[6] o diferentes tipos de aneuploidías (esto es anormalidades numéricas o estructurales en los cromosomas)[7] que por mutaciones en las mismas. El cáncer ha sido considerado también como una enfermedad metabólica, en la cual el metabolismo celular del oxígeno ha sido derivado de la ruta que genera energía (fosforilación oxidativa) hacia la vía que genera especies reactivas de oxígeno (ver la figura). Esto causa una conmutación de la vía principal de obtención de energía de la fosforilación oxidativa a la glucólisis anaeróbica (efecto conocido como efecto Warburg) y por consiguiente conlleva la acumulación de especies reactivas de oxígeno, lo cual conduce a su vez a estrés oxidativo que puede dañar el material genético de la célula desembocando finalmente en el cáncer (teoría del cáncer por estrés oxidativo).[8] Todas estas teorías de hecho pueden ser consideradas complementarias de las teorías clásicas, antes que teorías alternativas.
Otra teoría sobre el origen del cáncer fue desarrollada por astrobiólogos, y sugiere que el cáncer es, en realidad, un atavismo; es decir un retroceso evolutivo hacia una forma más primitiva de vida pluricelular.[9] Los genes responsables del crecimiento descontrolado de las células cancerosas son muy similares a algunos genes que se encuentran activos en algunos organismos pluricelulares muy primitivos y que les permitieron a estas primeras células agruparse y florecer. Estos genes aún existen en lo profundo del genoma de organismos más complejos, como los seres humanos; aunque algunos genes de desarrollo más reciente los mantienen bajo control. Cuando estos genes más recientes fallan en su misión de control (sea por la razón que sea), la célula se retrotrae a su programa de reproducción más primitivo y se reproduce fuera de control. Esta teoría es una alternativa a aquella teoría de que los cánceres comienzan con algunas células rebeldes que luego sufren un proceso de selección dentro del organismo. En esta alternativa las células rebeldes poseen tan solo un pequeño número de genes primitivos que pueden ser activados progresivamente, por lo que la variabilidad posible (los diferentes tipos de cánceres posibles), es relativamente muy limitada.[10]
Biología celular del cáncer
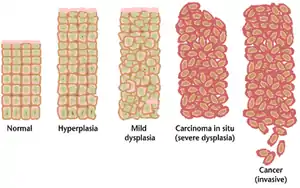
A menudo, los múltiples cambios genéticos que desembocan en la aparición del cáncer tardan años en acumularse. A lo largo de este tiempo, el comportamiento biológico de las células premalignas cambia suavemente de las propiedades de las células normales, a aquellas que son típicas de las células cancerosas. El tejido pre maligno presenta un aspecto característico bajo el microscopio. Entre estas características distintivas se encuentran un aumento del número de células en división, cambios en el tamaño y forma del núcleo celular, variaciones en el tamaño y forma de las propias células, pérdida de aquellas características que hacen a la diferenciación y especialización celular, y pérdida de la normal organización del tejido. La displasia por ejemplo, es un tipo anormal de proliferación celular excesiva caracterizada por la pérdida de la organización normal del tejido, y de la estructura celular en células premalignas. Estos cambios neoplásicos tempranos pueden ser distinguidos de la hiperplasia, que es un cambio reversible en la división celular causado por un estímulo externo, tal como un desbalance hormonal o una irritación crónica.
Los casos más severos de displasia son referidos como carcinomas in situ, es decir refiere a un carcinoma que a pesar de que ya está experimentando un crecimiento descontrolado, el grupo de células neoplásicas aún permanece en su ubicación original y no ha invadido otros tejidos. Sin embargo, un carcinoma in situ puede desarrollar una forma invasiva de malignidad, por lo que si es posible suele ser removido quirúrgicamente.
Evolución clonal
Al igual que una población de animales, una población de células cancerosas se encuentra sometida a una presión seleccionadora que eventualmente causará la muerte de aquellas células con características menos favorables para su supervivencia, este hecho sumado al proceso de mutación accidental que acompaña a todo proceso de replicación de material genético como generador de variedad; provoca que las células cancerosas realmente evolucionen dentro de un organismo por medio de un mecanismo que es en todo análogo al de selección natural. Este proceso indeseable es llamado evolución somática, y es debido a esto que el cáncer se torna más maligno.[11]
La mayor parte de los cambios en el metabolismo que habilita a las células a crecer en un patrón desordenado conducen también a la muerte celular. Sin embargo, una vez que el cáncer comienza, las células transformadas entran en un proceso de selección natural, en la cual aquellas pocas células que presentan cambios genéticos que aumentan sus posibilidades de supervivencia o reproducción continúan replicándose, y muy pronto se convierten en el tipo dominante en el tumor en crecimiento; mientras que por otra parte las células con cambios genéticos menos favorables quedan fuera de la competencia.[12] Este es un proceso en todo similar a aquel por el cual algunos patógenos tales como el SAMR se tornan resistentes a los antibióticos (o como el VIH obtiene resistencia a los antiretrovirales), y es la misma razón por la cual insectos y alimañas se tornan resistentes a los pesticidas. Este proceso de evolución somática es el motivo por el cual las recurrencias del cáncer pueden mostrar resistencia a los agentes quimioterápicos, y hasta en algunos casos, resistencia a la radioterapia.
Propiedades biológicas de las células cancerosas
En un artículo del año 2000 escrito por Hanahan y Weingerg, las propiedades biológicas de los tumores malignos fueron descritas como sigue:[13]
- Adquisición de autosuficiencia en cuanto a señales de crecimiento, conduciendo a un crecimiento descontrolado.
- Pérdida de sensibilidad a factores reguladores de crecimiento, conduciendo también a un crecimiento descontrolado.
- Pérdida de la capacidad de entrar en apoptosis, pudiendo de esta forma crecer a pesar de daños en el material genético y de señales anti crecimiento externas.
- Pérdida de la capacidad de senescencia, conduciendo esto a un potencial replicativo sin límites (literalmente a la inmortalidad).
- Adquisición de una capacidad sostenida de angiogenesis, permitiéndole al tumor crecer más allá de las limitaciones impuestas por la difusión pasiva de nutrientes.
- Adquisición de la capacidad de invadir los tejidos vecinos, es decir la característica que define a un carcinoma invasivo.
- Adquisición de la capacidad de producir metástasis en sitios distantes, una propiedad clásica de los tumores malignos (carcinomas y otros tipos de tumores).
El que estos múltiples pasos de desarrollo partiendo de una célula normal hasta llegar a una célula cancerosa sería un evento extremadamente fortuito sin:
- Pérdida en la capacidad de reparación de errores genéticos, conduciendo esto a una tasa de mutaciones aumentada (lo que se conoce como inestabilidad genómica), consiguiendo de este modo acelerar la aparición de los restantes cambios.
Este tipo de cambios biológicos son clásicos en los carcinomas; otro tipo de tumores malignos tales como los sarcomas pueden no necesitar alcanzarlos a todos. Por ejemplo, la invasión de tejidos y la capacidad de colonizar sitios distantes son propiedades normales de los leucocitos; por lo tanto estas capacidades no son un paso obligado en el desarrollo de la leucemia. Estos diferentes pasos, o capacidades adquiridas, no tienen que ser necesariamente representativos de mutaciones individuales. Por ejemplo, la inactivación de un único gen, el que codifica para la proteína p53, causa inestabilidad genómica, evasión del mecanismo de apoptosis y un incremento en la capacidad de angiogénesis. No todas las células en un cáncer se encuentran en proceso de división, en cambio, un subgrupo de estas llamadas, son las que se encuentran en proceso de división y son las que generan las células diferenciadas.[14]
Mecanismos
El cáncer es una enfermedad genética: para que las células comiencen a dividirse descontroladamente, se tiene que producir un daño en los genes que regulan el crecimiento.[15] Los protooncogenes son genes que promueven el crecimiento celular y la mitosis, mientras que los genes supresores de tumores desalientan el crecimiento celular; o provocan una parada momentánea en la división celular para llevar a cabo una reparación del ADN. Típicamente se requiere una serie de varias mutaciones afectando estos genes antes de que una célula normal pueda transformarse en una célula cancerosa. Este concepto es algunas veces referido como oncoevolución. Las mutaciones en estos genes proveen a los tumores de las señales necesarias para que las células cancerosas comiencen a dividirse en forma descontrolada. Sin embargo, la división celular descontrolada que caracteriza al cáncer también requiere que las células en división sean capaces de duplicar todos los genes y componentes celulares mínimos necesarios para crear células hijas funcionales. La activación del mecanismo de glicólisis anaeróbica (el efecto Warburg), el cual no necesariamente es inducido por mutaciones ni en los protooncogenes ni en los genes supresores de tumores,[16] provee la mayor parte de los bloques de construcción necesarios para duplicar los componentes celulares en una célula en división y es, por lo tanto, esencial en el proceso de oncogénesis.[8]
Tipos celulares involucrados en el crecimiento tumoral
Hay varios tipos celulares diferentes que resultan críticos para el crecimiento tumoral. En particular las células progenitoras endoteliales son una población celular muy importante en el crecimiento de los vasos sanguíneos asociados a los tumores.[17][18] La hipótesis de que las células progenitoras endoteliales son importantes en el crecimiento tumoral, angiogénesis y metástasis ha sido recientemente apoyada por una investigación publicada en Cancer Research (de agosto de 2010). Esta publicación sostiene que las células progenitoras endoteliales pueden ser marcadas utilizando el Inhibidor de Unión del ADN 1 (ID1 por sus siglas en inglés). Este nuevo hallazgo implica que los investigadores pueden ser capaces de rastrear el movimiento de células progenitoras desde su origen en la médula ósea, su pasaje hacia la circulación sanguínea y de allí hacia el estroma del tumor y su vasculatura. Este hallazgo de que las células progenitoras endoteliales son de hecho incorporadas activamente en la vasculatura del tumor brinda evidencia de la importancia de este tipo celular en el desarrollo de la vasculatura que soporta al tumor y en la metástasis. Y aún más, la ablación de células progenitoras endoteliales en la médula ósea conduce a una significativa disminución en el crecimiento tumoral y en el desarrollo de vasculatura. La continuación de investigaciones en relación con la importancia de las células progenitoras endoteliales puede representar el descubrimiento de nuevos blancos terapéuticos en el tratamiento de algunos tipos de cáncer.[19]
Oncogenes
Los oncogenes promueven el crecimiento celular a través de una gran variedad de mecanismos. Muchos son capaces de producir hormonas, es decir "mensajeros químicos" que comunican diferentes células y que pueden promover la mitosis, siendo el efecto de las mismas dependiente de la transducción de señales típica del tejido que las recibe o de las células que lo componen. En otras palabras, cuando un receptor hormonal presente en la célula receptora es estimulado, la señal recibida en la superficie de la célula es conducida al núcleo para producir algunos cambios en la regulación de la transcripción de genes a nivel nuclear. Algunos oncogenes forman parte por sí mismos del sistema de transducción de señales, o codifican los receptores encargados de recibirlas en las células y tejidos, controlando de esta manera la sensibilidad de los mismos a diferentes hormonas. Muchos oncogenes producen mitógenos, o se encuentran involucrados en la transcripción genética del ADN durante la biosíntesis de las proteínas responsables de diferentes productos utilizados por las células en su interacción con otras células.
Las mutaciones en los protooncogenes, los cuales son los homólogos normalmente en reposo de los oncogenes, pueden modificar su nivel de expresión y función, aumentando la actividad o la cantidad de producto proteico. Cuando esto ocurre, los protooncogenes se transforman en oncogenes, y esta transición altera el balance normal del mecanismo de regulación del ciclo celular, haciendo posible un crecimiento descontrolado.
Sin embargo, no es posible reducir la probabilidad de que se produzca un cáncer removiendo los protooncogenes del genoma de un organismo, ya que estos genes son críticos para el crecimiento, reparación y homeostasis del organismo. Y solo cuando se encuentran mutados es cuando las señales que impulsan el crecimiento se tornan excesivas.
Uno de los primeros oncogenes en ser definidos por la investigación en el área del cáncer fue el gen Ras. Las mutaciones en los protooncogenes de la familia Ras (la cual comprende al H-Ras, N-Ras y K-Ras) son muy comunes, siendo encontradas entre el 20 al 30 % de todos los tumores humanos.[20] El gen Ras fue originalmente identificado en el genoma del virus del sarcoma de Harvey, y los investigadores se encontraron con la sorpresa de que este gen no solo se encuentra presente en el genoma humano, sino que además, cuando se encuentra ligado a elementos que controlan la estimulación, es capaz de inducir el cáncer en líneas celulares en cultivo.[21]
Protooncogenes
Los protooncogenes promueven el crecimiento celular en una gran variedad de formas. Muchos de estos genes pueden producir hormonas —mensajeros químicos entre células— que alientan la mitosis, el efecto de las cuales va a depender del mecanismo de transducción de señales y de la bioquímica particular de cada receptor en las células y tejidos diana, controlando de esta forma la sensibilidad a tales hormonas.
Muchos protooncogenes producen mitógenos, o se encuentran implicados en la transcripción del ADN durante la síntesis de proteínas que es la responsable de producir las proteínas y enzimas que las células utilizan para interactuar entre sí y para la maquinaria de síntesis de compuestos químicos.
Las mutaciones ocurridas en los protooncogenes pueden modificar su potencial de expresión génica y funcionalidad, aumentando ya sea la cantidad o la actividad de la proteína producida. Cuando esto ocurre, estos protooncogenes se convierten en oncogenes, y las células que los poseen adquieren una alta probabilidad de dividirse en forma excesiva o descontrolada. La probabilidad de que se produzca un cáncer no puede ser reducida removiendo los protooncogenes del genoma, ya que estos desempeñan una función crítica en el crecimiento, reparación y homeostasis del organismo. Y es solo cuando aparece una mutación en alguno de estos genes que las señales de crecimiento se tornan excesivas.
Es importante destacar que un gen que posee un rol promotor del crecimiento puede incrementar el potencial carcinogénico de una célula, bajo la condición de que todos los mecanismos necesarios para permitir el crecimiento celular se encuentren activados.[22] Esta condición incluye también la activación de genes supresores tumorales específicos (ver más abajo). Si estas condiciones no se cumplimentan, la célula puede dejar de crecer y entrar en un proceso que culmina con su muerte. Esto hace que el conocimiento de los oncogenes que regulan cada una de las etapas y tipos de células cancerosas, sea algo crucial para el desarrollo de nuevas estrategias de tratamiento.
Genes supresores tumorales
Los genes supresores tumorales codifican para proteínas y señales antiproliferación que suprimen la mitosis y el crecimiento celular. Generalmente, los genes supresores tumorales son factores de transcripción que se activan por mecanismos de estrés celular o daño en el ADN. A menudo el daño en el ADN produce la aparición de material genético flotando libre en el interior celular y otros signos, lo cual desencadena la activación de enzimas y vías metabólicas que conducen a la activación de los genes supresores tumorales. La función de estos genes es detener la progresión del ciclo celular con el objeto de llevar a cabo una reparación del material genético, previniendo de esta forma que las posibles mutaciones sean pasadas a las células hijas. La proteína p53, producida por uno de los más estudiados genes de supresión tumoral, es un factor de transcripción activado por muchos factores de estrés celular, entre los que se incluyen la hipoxia, y el daño por radiación ultravioleta.
A pesar de que aproximadamente la mitad de todos los cánceres posiblemente involucran una mutación en el gen p53, la función supresora de este gen todavía es pobremente entendida. La proteína p53 tiene claramente dos funciones: por un lado cumple un rol en el núcleo celular como factor de transcripción, y por otro lado cumple una función citoplasmática regulando el ciclo y división celulares y la apoptosis.
La hipótesis de Warburg hace referencia al uso preferencial de la glicólisis como mecanismo de obtención de energía para sostener el crecimiento canceroso. El gen p53 ha demostrado regular el cambio entre la vía respiratoria y la glicolítica.[23]
Sin embargo, una mutación puede por sí misma, dañar al propio gen supresor, o a la señal que lo activa, "apagándolo". La invariable consecuencia de esto es que el mecanismo de reparación del ADN resulta dañado o inhibido. Las mutaciones en el ADN se acumulan sin reparación, conduciendo inevitablemente al cáncer.
Las mutaciones en los genes supresores tumorales que ocurren en las células de línea germinal son pasadas a la descendencia, aumentando la probabilidad de aparición de cáncer en las generaciones subsiguientes. Los miembros de estas familias presentan una incidencia aumentada y una latencia disminuida en la aparición de tumores múltiples. Los tipos de tumores involucrados son típicos de cada tipo de mutación en cada gen supresor involucrado; con algunas mutaciones causando incluso tipos específicos de cáncer. El modo de herencia de los genes supresores tumorales causa que los miembros que heredan al menos una copia mutada del gen resultan afectados. Por ejemplo, los individuos que heredan el alelo mutado del gen p53 (y que por lo tanto resultan heterocigotas para la mutación) pueden desarrollar melanomas y cáncer de páncreas, condición conocida como síndrome de Li-Fraumeni. Otros síndromes causados por genes supresores tumorales mutados incluyen las mutaciones en la proteína del retinoblastoma (Rb), y la vinculada a la poliposis adenomatosa familiar que predispone al cáncer de colon. El cáncer de colon por adenopoliposis se encuentra asociado a cientos de pólipos en el colon cuando el individuo es joven, conduciendo al cáncer colorrectal a edades relativamente tempranas. Finalmente, las mutaciones heredadas en los genes BRCA1 y BRCA2 conducen a la aparición temprana del cáncer de mama.
En 1971 Knudson propuso que el desarrollo del cáncer dependía de al menos dos eventos de mutación, razón por la cual se conoció a esta hipótesis como hipótesis de Knudson o hipótesis de los dos eventos; sin embargo, una mutación en un gen supresor de línea germinal puede causar cáncer en las personas que lo heredan con tan solo un evento más de mutación ocurrido en forma más tardía en la vida del organismo, que inactive el otro alelo de ese gen.[24]
Por lo general, los oncogenes son dominantes, ya que suelen presentar mutaciones con ganancia de función, mientras que los genes supresores tumorales son genes recesivo, ya que por definición suelen contener mutaciones con pérdida de función. Cada célula posee dos copias del mismo gen, una de cada progenitor, y en la mayor parte de los casos una mutación con ganancia de función en tan solo una copia de un protooncogén en particular es suficiente para convertir a ese protooncogén en un verdadero oncogén. Por otro lado, las mutaciones con pérdida de función tienen que ocurrir en ambas copias del gen para que la actividad del mismo desaparezca por completo. Sin embargo, existen casos en los cuales una única copia mutada de un gen supresor tumoral puede provocar la pérdida total de función, por un mecanismo que lleva a que la copia silvestre del gen se torne no funcional. Este fenómeno es conocido como efecto dominante negativo, y puede ser observado en muchas mutaciones del gen p53.
El modelo de dos eventos de Knudson, ha sido recientemente cuestionado por varios investigadores. La inactivación de un alelo en algunos genes supresores tumorales es suficiente para causar tumores. Este fenómeno es conocido como haplosuficiencia y ha sido demostrado por numerosas investigaciones basadas en diferentes enfoques. Los tumores causados por haplosuficiencia tienen, por lo general, una edad de aparición tardía al ser comparados por aquellos tumores causados por un proceso de dos eventos.[25]
Mutaciones múltiples
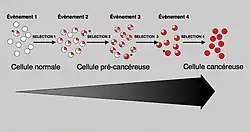
En general se requiere que haya mutaciones en ambos tipos de genes para que el cáncer pueda ocurrir. Por ejemplo, una mutación limitada a un único oncogén, puede ser suprimida por los genes supresores tumorales y por el proceso de control de la mitosis normal. Esto fue hipotetizado por primera vez por Knudson.[26] Una mutación en un único gen supresor tumoral, tampoco causa cáncer, debido a la presencia de muchos genes de respaldo que duplican sus funciones. Y es solo cuando un buen número de protooncogenes han mutado a oncogenes, y cuando un número lo suficientemente grande de genes supresores se han desactivado o dañado, que las señales de crecimiento celular sobrepasan a las señales de regulación causando el crecimiento descontrolado de la célula. Muy a menudo, la tasa de mutaciones aumenta a medida que la célula envejece, debido a que los propios genes que previenen las mutaciones resultan dañados; es por esto que el daño al ADN forma un bucle de retroalimentación.
Por lo general los oncogenes son alelos dominantes, ya que contienen mutaciones con ganancia de función, mientras que los genes supresores tumorales son alelos recesivos, ya que contienen mutaciones con pérdida de función. Cada célula contiene dos copias de cada gen, uno proveniente de cada progenitor, y, en la mayor parte de los casos, la ganancia de función en solo una copia de algún protooncogén en particular es suficiente para convertir a uno de estos protooncogenes en un verdadero oncogén; mientras que por lo general las mutaciones con pérdida de función tienen que ocurrir en ambas copias de un gen supresor tumoral para que se vea una pérdida total de función. Sin embargo, existen algunos casos en los cuales la pérdida de función de tan solo una copia de un gen supresor tumoral puede inducir a la otra copia a dejar de funcionar, a esto se le llama efecto dominante negativo. Esto puede ser observado en muchas mutaciones del gen p53.
Las mutaciones que ocurren en los genes supresores, pero no de cualquier célula, sino de células de línea germinal; pueden causar un aumento heredable en la probabilidad de sufrir algunos tipos de cáncer. Los miembros pertenecientes a estas familias poseen una incidencia aumentada de estos tipos de cáncer y una latencia menor (transcurre menos tiempo hasta que aparecen tumores múltiples). El modo de herencia que caracteriza a los genes supresores tumorales mutados es que afecta a aquellos miembros que heredan una copia defectuosa de uno de sus progenitores y una copia normal del otro. Esto es debido a que las mutaciones en los genes supresores tumorales actúan de forma recesiva —hay, sin embargo, algunas excepciones a esta regla—, la pérdida de función del gen normal provoca la aparición del fenotipo canceroso. Por ejemplo, los individuos que son heterocigotas para las mutaciones del gen p53 a menudo son víctimas del síndrome de Li-Fraumeni, y aquellos que son heterocigotas para el gen Rb desarrollan retinoblastoma. De forma similar las mutaciones en el gen poliposis adenomatosa familiar se encuentran ligadas a cáncer colorrectal con presencia de múltiples pólipos en el colon cuando el paciente aún es joven. Mientras que las mutaciones en los genes BRCA1 y BRCA2 conducen a una aparición temprana de cáncer de mama.
Una versión extrema del proceso de múltiples mutaciones es la hipótesis llamada cromotripsis (composición que hace referencia al proceso de ruptura —tripsis— de un cromosoma). Según esta hipótesis, en algo así como un 2-3 % de todos los casos de cáncer y algo más de un 25 % de los cánceres de hueso, se produce un despedazamiento catastrófico de un cromosoma en decenas o cientos de pedazos luego de lo cual la maquinaria de reparación del ADN los recompone, pero de forma incorrecta. Este despedazamiento se produce probablemente mientras los cromosomas se compactan antes de la división celular, aunque el detonante de este proceso es aún desconocido. Bajo este modelo, la aparición del cáncer es el resultado de un único evento aislado, antes que la acumulación de múltiples mutaciones.[27]
Carcinógenos no mutagénicos
Muchos mutágenos son también carcinógenos, pero algunos carcinógenos no son mutágenos. Entre estos últimos tenemos algunos ejemplos tales como el alcohol y el estrógeno. Se cree que estas sustancias promueven la aparición de cáncer a través del efecto estimulante en la división celular que producen. A mayores tasas de mitosis, se produce una disminución en las oportunidades que tienen las enzimas de reparación del ADN de corregir los errores antes de que se asienten definitivamente luego de replicación del ADN, incrementando de esta forma la probabilidad de aparición de un fallo genético. De la misma forma un error durante la mitosis puede conducir a que las células hijas reciban un número erróneo de cromosomas, lo que produce aneuploidía y puede provocar cáncer.
Bacterianas
Por ejemplo la bacteria Helicobacter pylori es conocida por causar linfoma MALT. Otros tipos de bacterias han sido implicadas en otros cánceres.
Virales
Muchos tipos de cáncer tienen su origen en una infección viral; esto resulta especialmente cierto en algunos animales tales como las aves, pero resulta algo menos frecuente en humanos. Algo así como un 12 % de todos los tipos de cánceres en humanos pueden ser atribuidos a infecciones virales.[28] Las modalidades de los tumores inducidos por virus pueden dividirse en dos grandes categorías, transformación aguda o transformación lenta. En los virus de transformación aguda, la partícula viral acarrea un gen que actúa in-vivo como un oncogén hiperactivo llamado oncogén viral (v-onc) y la célula infectada se transforma tan pronto como comienza la expresión del v-onc. En contraste, los virus de transformación lenta requieren que el genoma viral sea insertado en el genoma de la célula hospedadora —este paso de inserción viral es obligado en los retrovirus— en una región próxima a un protooncogén del genoma hospedador. El promotor génico del virus o algún otro elemento de regulación causa la sobreexpresión del protooncogén presente en el genoma hospedador, el cual, induce a su vez una proliferación celular descontrolada. Debido a que la inserción del genoma viral no es específica ni tiene tropismo por ningún protooncogén en particular, la chance de que se inserte en una región cercana a un protooncogén es baja, por lo que los virus de transformación lenta tienen un período de latencia hasta la aparición del tumor mucho más largo si se lo compara con los virus de transformación aguda, los cuales de hecho llevan su propio oncogén viral.
Algunos virus que se sabe que son capaces de provocar cáncer son el HPV (causante de cáncer de cuello de útero) HBV (causante de cáncer de hígado), y el EBV (que puede provocar un tipo de linfoma). Todos ellos son virus de ADN. Se cree que cuando un virus infecta una célula, inserta una parte de su propio ADN cerca de los genes de crecimiento de la propia célula, causando división celular. El grupo de células modificadas se forma a partir de la primera célula en división que posee el mismo ADN viral en la región cercana a los genes de crecimiento. Este grupo de células modificadas, resultan entonces especiales, debido a que al menos uno de sus mecanismos de control normales se ha perdido.
Dependiendo de su localización, las células pueden resultar dañadas por la radiación solar, sustancias químicas provenientes del humo del cigarrillo, o inflamación a causa de bacterias o virus. Cada célula que sufre un daño, es un paso más en la vía que conduce al cáncer. Las células a menudo mueren si resultan demasiado dañadas, ya sea a través de un fallo en sus procesos vitales o a causa del sistema inmune; sin embargo, a veces el cambio noquea tan solo a un único gen causante de cáncer. En una persona añosa, existen cientos, decenas de cientos o miles de células noqueadas. La chance de que alguna de ellas se convierta en cancerosa es muy baja.
Cuando ocurre un daño en cualquier área de una célula transformada, algo diferente ocurre. Cada una de ellas tiene el potencial de crecimiento. Las células transformadas se dividen mucho más rápido cuando el área resulta dañada por agentes físicos, químicos o infecciosos. Se establece un círculo vicioso: el daño en el área causa la división de células transformadas, causando una mayor probabilidad de que sufran posteriores daños en genes críticos.
Este modelo de carcionogénesis es popular debido a que explica por qué los cánceres crecen. Se podría esperar que las células que resultan dañadas debido a radiación muriesen o al menos tuviesen menos chances de sobrevivir debido a que poseen menos genes funcionales; los virus incrementan en número de genes en funcionamiento.
Una preocupación es que podríamos terminar con cientos de vacunas destinadas a prevenir la infección por cada uno de los virus que pueden modificar nuestras células. Sin embargo, los virus pueden tener diferentes efectos en diferentes partes del organismo. Por lo que resulta posible prevenir un cierto número de diferentes tipos de cáncer inmunizando contra un único agente viral. Por ejemplo, es bastante probable que el HPV desempeñe un rol en el desarrollo de algunos cánceres de la mucosa oral, además de su capacidad reconocida sobre la mucosa del cérvix uterino.
Helmintiasis
Ciertos gusanos parasíticos son conocidos por tener capacidad carcinogénica.[29] These include:
- Clonorchis sinensis (el organismo causante de Clonorquiasis) y Opisthorchis viverrini (causante de Opistorquiasis) presentan una asociación con la aparición de colangiocarcinoma.[30]
- Varias especies de Schistosoma (los organismos causantes de esquistosomiasis) se encuentran relacionados al cáncer de vejiga.
Epigenética
Epigenética es el estudio de la regulación en la expresión de los genes a través de modificaciones químicas, que sin ser mutaciones, afectan al ADN. La teoría epigenética en la patogénesis del cáncer es que estos cambios no mutacionales en el ADN pueden conducir a alteraciones en la expresión de los genes. Normalmente los oncogenes son silenciosos, por ejemplo, debido a metilación del ADN. La pérdida de esa metilación puede inducir a la expresión aberrante de oncogenes, desembocando en la patogénesis del cáncer. Los mecanismos conocidos de control epigenético incluyen a la metilación del ADN, y la metilación o acetilación de las histonas que son proteínas unidas al ADN cromosomal en lugares específicos. Algunos tipos de drogas tales como los inhibidores HDAC y los inhibidores de ADN metiltransferasa, pueden contribuir a la regular nuevamente la señalización epigenética en la célula cancerosa.
Células madre del cáncer
Una nueva forma de ver al proceso de oncogénesis proviene de integrar las ideas de la biología evolutiva con las de la oncología. La hipótesis de la célula madre del cáncer propone que los diferentes tipos de células que se encuentran en un tumor heterogéneo provienen de una única célula, llamada célula madre del cáncer. Estas células madre cancerosas pueden provenir de la transformación de las células adultas o diferenciadas del organismo. Estas células madre persisten como un subcomponente del tumor y retienen algunas propiedades clave de célula madre. Estas células pueden producir por diferenciación una gran cantidad de células diferentes, son capaces de autorrenovación y control homeostático.[31] Además, la aparición de recaídas del cáncer y metástasis también se atribuyen a estas células. La hipótesis de las células madre del cáncer no contradice los conceptos anteriormente descritos de la oncogénesis.
Evolución clonal
Mientras que los cambios genéticos y epigenéticos en los genes supresores tumorales cambian el comportamiento de las células, estas alteraciones, al fin, resultan en cáncer a través de sus efectos en la población de células neoplásicas y su microambiente.[32] Las células dentro de una neoplasia compiten por espacio y recursos. Por lo tanto un clon con una mutación en un gen supresor tumoral o en un oncogen podrá expandirse dentro de la neoplasia únicamente si esa mutación le brinda una ventaja competitiva sobre los demás clones y células normales presentes en su microambiente.[33] Es por esto que el proceso de oncogénesis es, formalmente, un proceso de evolución Darwiniana, conocido como evolución somática o clonal.[34] Además, bajo la luz de los mecanismos Darwinianos de oncogénesis, se ha teorizado que las diferentes formas de cáncer pueden ser categorizadas como puberales y gerontológicas. Las investigaciones antropológicas que están siendo conducidas actualmente
La investigación antropológica que se está realizando actualmente con respecto al cáncer como un proceso evolutivo natural, a través del cual la selección natural destruye aquellos fenotipos que en relación con su ambiente resultan competitivamente inferiores; mientras que favorece a otros.
Según esta teoría, el cáncer se presenta en dos tipos distintos: Por un lado aquel que se presenta desde el nacimiento hasta el final de la pubertad (aproximadamente a los 20 años) teleológicamente inclinado hacia una dinámica de soporte en grupo, y desde mediados de la vida hasta la muerte (desde los 40 años en adelante) teleológicamente inclinado en dirección contraria a una dinámica de sobrepoblación.[cita requerida]
Referencias
- García, Martín Granados; Rodríguez, Oscar Arrieta; León, David Cantú de (16 de noviembre de 2012). Oncología y cirugía. Bases y principios. Editorial El Manual Moderno. ISBN 9786074482874. Consultado el 19 de marzo de 2018. «Los agentes etiológicos son las causas directas de la transformación maligna, ya que alteran diversos mecanismos genéticos y bioquímicos. A este último proceso se le conoce como carcinogénesis u oncogénesis. »
- Croce CM (January de 2008). «Oncogenes and cancer». The New England Journal of Medicine 358 (5): 502-11. PMID 18234754. doi:10.1056/NEJMra072367.
- Knudson AG (November de 2001). «Two genetic hits (more or less) to cancer». Nature reviews. Cancer 1 (2): 157-62. PMID 11905807. doi:10.1038/35101031.
- Fearon ER, Vogelstein B (June de 1990). «A genetic model for colorectal tumorigenesis». Cell 61 (5): 759-67. PMID 2188735. doi:10.1016/0092-8674(90)90186-I.
- Villeneuve, PJ; Mao Y (November de 1994). «Lifetime probability of developing lung cancer, by smoking status, Canada». Canadian Journal of Public Health 85 (6): 385-388. PMID 7895211.
- Jaffe, LF (2003). «Epigenetic theories of cancer initiation». Advances in cancer research. Advances in Cancer Research 90: 209-30. ISBN 978-0-12-006690-2. PMID 14710952. doi:10.1016/S0065-230X(03)90007-8.
- Rasnick, D; Duesberg, PH (1999). «How aneuploidy affects metabolic control and causes cancer». The Biochemical journal 340 (3): 621-30. PMC 1220292. PMID 10359645. doi:10.1042/0264-6021:3400621.
- López-Lázaro, M (2010). «A New View of Carcinogenesis and an Alternative Approach to Cancer Therapy». Molecular medicine 16 (3–4): 144-153. PMC 2802554. PMID 20062820. doi:10.2119/molmed.2009.00162.
- P C W Davies and C H Lineweaver (February de 2011). «Cancer tumors as Metazoa 1.0: tapping genes of ancient ancestors». Phys. Biol. 8 (1): 1-7. doi:10.1088/1478-3975/8/1/015001.
- Dean, Tim. "Cancer resembles life 1 billion years ago, say astrobiologists", Australian Life Scientist, 08 February 2011. Retrieved on 2011-02-15.
- Nowell PC (October de 1976). «The clonal evolution of tumor cell populations». Science 194 (4260): 23-8. PMID 959840. doi:10.1126/science.959840.
- Merlo LM, Pepper JW, Reid BJ, Maley CC (December de 2006). «Cancer as an evolutionary and ecological process». Nat Rev Cancer 6 (12): 924-35. PMID 17109012. doi:10.1038/nrc2013.
- Hanahan D, Weinberg RA (2000). «The hallmarks of cancer». Cell 100 (1): 57-70. PMID 10647931. doi:10.1016/S0092-8674(00)81683-9.
- Cho RW, Clarke MF (February de 2008). «Recent advances in cancer stem cells». Curr. Opin. Genet. Dev. 18 (1): 48-53. PMID 18356041. doi:10.1016/j.gde.2008.01.017.
- Vogelstein, Bert; Kinzler, Kenneth W (2004). «Cancer genes and the pathways they control». Nature Medicine 10 (8): 789-99. PMID 15286780. doi:10.1038/nm1087.
- Brand, KA; Hermfisse, U (1997). «Aerobic glycolysis by proliferating cells: a protective strategy against reactive oxygen species». The FASEB Journal 11 (5): 388-95. PMID 9141507.
- Gao D et al. (2008). «Endothelial Progenitor Cells Control the Angiogenic Switch in Mouse Lung Metastasis». Science 319 (5860): 195-198. PMID 18187653. doi:10.1126/science.1150224.
- Nolan DJ et al. (2007). «Bone marrow-derived endothelial progenitor cells are a major determinant of nascent tumor neovascularization». Genes & Development 21 (12): 1546-1558. PMC 1891431. PMID 17575055. doi:10.1101/gad.436307.
- Mellick As, Plummer PN et al. (2010). «Using the Transcription Factor Inhibitor of DNA Binding 1 to Selectively Target Endothelial Progenitor Cells Offers Novel Strategies to Inhibit Tumor Angiogenesis and Growth». Cancer Research 70 (18): 7273-7282. PMC 3058751. PMID 20807818. doi:10.1158/0008-5472.CAN-10-1142.
- Bos JL (September de 1989). «ras oncogenes in human cancer: a review». Cancer Research 49 (17): 4682-9. PMID 2547513. Consultado el 6 de junio de 2009.
- Chang EH, Furth ME, Scolnick EM, Lowy DR (1982). «Tumorigenic transformation of mammalian cells induced by a normal human gene homologous to the oncogene of Harvey murine sarcoma virus». Nature 297 (5866): 479-83. PMID 6283358. doi:10.1038/297479a0.
- Vlahopoulos SA, Logotheti S, Mikas D, Giarika A, Gorgoulis V, Zoumpourlis V (April de 2008). «The role of ATF-2 in oncogenesis». BioEssays 30 (4): 314-27. PMID 18348191. doi:10.1002/bies.20734.
- Matoba S, Kang J, Patino W, Wragg A, Boehm M, Gavrilova O, Hurley P, Bunz F, Hwang P (2006). «p53 regulates mitochondrial respiration». Science 312 (5780): 1650-3. PMID 16728594. doi:10.1126/science.1126863.
- Knudson A (1971). «Mutation and Cancer: Statistical Study of Retinoblastoma». Proc Natl Acad Sci USA 68 (4): 820-3. PMC 389051. PMID 5279523. doi:10.1073/pnas.68.4.820.
- Fodde R, Smits R (2002). «Cancer biology. A matter of dosage». Science 298 (5594): 761-3. PMID 12399571. doi:10.1126/science.1077707.
- Knudson AG (November de 2001). «Two genetic hits (more or less) to cancer». Nature Reviews. Cancer 1 (2): 157-62. PMID 11905807. doi:10.1038/35101031.
- Stephens PJ, Greenman CD, Fu B, et al. (January de 2011). «Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development». Cell 144 (1): 27-40. PMC 3065307. PMID 21215367. doi:10.1016/j.cell.2010.11.055. Resumen divulgativo – The New York times (10 January 2011).
- Carrillo-Infante C, Abbadessa G, Bagella L, Giordano A (June de 2007). «Viral infections as a cause of cancer (review)». Int. J. Oncol. 30 (6): 1521-8. PMID 17487374.
- Safdar, Amar (1 de junio de 2011). Management of Infections in Cancer Patients. Springer. pp. 478-. ISBN 978-1-60761-643-6. Consultado el 17 de agosto de 2011.
- Samaras V, Rafailidis PI, Mourtzoukou EG, Peppas G, Falagas ME (May de 2010). «Chronic bacterial and parasitic infections and cancer: a review». J Infect Dev Ctries 4 (5): 267-81. PMID 20539059.
- Dalerba, P.; Cho, R. W.; Clarke, M. F. (2007). «Cancer stem cells: models and concepts». Annu. Rev. Med. 58: 267-284. PMID 17002552. doi:10.1146/annurev.med.58.062105.204854.
- Nowell PC (October de 1976). «The clonal evolution of tumor cell populations». Science 194 (4260): 23-8. PMID 959840. doi:10.1126/science.959840.
- Zhang W, Hanks AN, Boucher K et al. (January de 2005). «UVB-induced apoptosis drives clonal expansion during skin tumor development». Carcinogenesis 26 (1): 249-57. PMC 2292404. PMID 15498793. doi:10.1093/carcin/bgh300.
- Merlo LM, Pepper JW, Reid BJ, Maley CC (December de 2006). «Cancer as an evolutionary and ecological process». Nature Reviews. Cancer 6 (12): 924-35. PMID 17109012. doi:10.1038/nrc2013.
Radoslav S.Jovic-Cancer-released ancestor from our genes, www.newcancertheory.com
Lecturas recomendadas
- Tokar, Erik J.; Benbrahim-Tallaa, Lamia; Waalkes, Michael P. (2011). «Chepter 14. Metal Ions in Human Cancer Development». En Astrid Sigel, Helmut Sigel and Roland K. O. Sigel, ed. Metal ions in toxicology: effects, interactions, interdependencies. Metal Ions in Life Sciences 8. RSC Publishing. pp. 375-401. doi:10.1039/9781849732116-00375.
- Dixon K, Kopras E (2004). «Genetic alterations and DNA repair in human carcinogenesis». Semin Cancer Biol 14 (6): 441-8. PMID 15489137. doi:10.1016/j.semcancer.2004.06.007.
- Kleinsmith, Lewis J (2006). Principles of cancer biology. San Francisco: Pearson Benjamin Cummings. ISBN 978-0-8053-4003-7.
- Sarasin A (2003). «An overview of the mechanisms of mutagenesis and carcinogenesis». Mutat Res 544 (2–3): 99-106. PMID 14644312. doi:10.1016/j.mrrev.2003.06.024.
- Schottenfeld D, Beebe-Dimmer JL (2005). «Advances in cancer epidemiology: understanding causal mechanisms and the evidence for implementing interventions». Annu Rev Public Health 26: 37-60. PMID 15760280. doi:10.1146/annurev.publhealth.26.021304.144402.
- Tannock, Ian; Hill, Richard; Bristow, Robert; Harrington, Lea (2005). The basic science of oncology (4th edición). New York: McGraw-Hill. ISBN 978-0-07-138774-3.
- Wicha MS, Liu S, Dontu G (2006). «Cancer stem cells: an old idea--a paradigm shift». Cancer Res 66 (4): 1883-90. PMID 16488983. doi:10.1158/0008-5472.CAN-05-3153.
| Control de autoridades |
|
|---|
 Datos: Q1637543
Datos: Q1637543 Multimedia: Carcinogenesis / Q1637543
Multimedia: Carcinogenesis / Q1637543