ChIP-on-chip
ChIP-on-chip (también conocido como ChIP-chip) es una tecnología que combina inmunoprecipitación de cromatina (ChIP) con micromatriz de ADN (chip). Al igual que ChIP normal, ChIP-on-chip se utiliza para investigar las interacciones entre las proteínas y el ADN in vivo. Específicamente, permite la identificación del cistrome, la suma de los sitios de unión, para las proteínas de unión al ADN en todo el genoma.[1] Se puede realizar un análisis del genoma completo para determinar las ubicaciones de los sitios de unión para casi cualquier proteína de interés.[1] Como sugiere el nombre de la técnica, tales proteínas son generalmente las que operan en el contexto de la cromatina. Los representantes más destacados de esta clase son los factores de transcripción, las proteínas relacionadas con la replicación, como la proteína del complejo de reconocimiento de origen (ORC), las histonas, sus variantes y las modificaciones de las histonas.
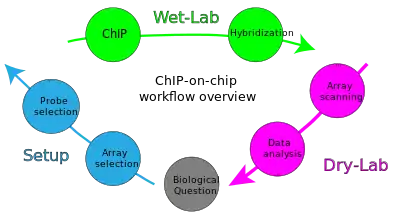
El objetivo de ChIP-on-chip es localizar sitios de unión a proteínas que puedan ayudar a identificar elementos funcionales en el genoma. Por ejemplo, en el caso de un factor de transcripción como proteína de interés, se pueden determinar sus sitios de unión al factor de transcripción en todo el genoma. Otras proteínas permiten la identificación de regiones promotoras, potenciadoras, represoras y elementos silenciadores, aislantes, elementos de frontera y secuencias que controlan la replicación del ADN.[2] Si las histonas son un tema de interés, se cree que la distribución de modificaciones y sus localizaciones pueden ofrecer nuevos conocimientos sobre los mecanismos de regulación.
Uno de los objetivos a largo plazo para los que se diseñó ChIP-on-chip es establecer un catálogo de organismos (seleccionados) que enumere todas las interacciones proteína-ADN en diversas condiciones fisiológicas. Este conocimiento ayudaría en última instancia a comprender la maquinaria detrás de la regulación génica, la proliferación celular y la progresión de la enfermedad. Por lo tanto, ChIP-on-chip ofrece potencial para complementar nuestro conocimiento sobre la orquestación del genoma a nivel de nucleótidos e información sobre niveles más altos de información y regulación a medida que se propaga por la investigación en epigenética.
Plataformas tecnológicas
Las plataformas técnicas para realizar experimentos ChIP-on-chip son las micromatrices de ADN, o "chips". Se pueden clasificar y distinguir según varias características:
- Tipo de sonda: las matrices de ADN pueden comprender cDNA o productos de PCR aplicados mecánicamente, oligonucleótidos aplicados mecánicamente u oligonucleótidos que se sintetizan in situ. Las primeras versiones de microarreglos se diseñaron para detectar ARN de regiones genómicas expresadas (marcos de lectura abiertos, también conocidos como ORF). Aunque dichas matrices son perfectamente adecuadas para estudiar los perfiles de expresión génica, tienen una importancia limitada en los experimentos de ChIP, ya que la mayoría de las proteínas "interesantes" con respecto a esta técnica se unen en regiones intergénicas. Hoy en día, incluso las matrices hechas a medida se pueden diseñar y ajustar para que coincidan con los requisitos de un experimento. Además, se puede sintetizar cualquier secuencia de nucleótidos para cubrir regiones génicas e intergénicas.
- Tamaño de la sonda: la versión anterior de las matrices de cDNA tenía una longitud de sonda de aproximadamente 200 pb. Las últimas versiones de matriz utilizan oligos desde 70- (Microarrays, Inc.) hasta 25-mers (Affymetrix).
- Composición de la sonda: Hay matrices de ADN en mosaico y sin mosaico. Las matrices sin teselas utilizan sondas seleccionadas según criterios no espaciales, es decir, las secuencias de ADN utilizadas como sondas no tienen distancias fijas en el genoma. Sin embargo, las matrices en mosaico seleccionan una región genómica (o incluso un genoma completo) y la dividen en partes iguales. Tal región se llama ruta en mosaico. La distancia promedio entre cada par de fragmentos vecinos (medida desde el centro de cada fragmento) da la resolución de la ruta en mosaico. Una ruta puede estar superpuesta, de extremo a extremo o espaciada.[3]
- Tamaño de la matriz: las primeras micromatrices utilizadas para ChIP-on-Chip contenían alrededor de 13 000 segmentos de ADN manchado que representaban todos los ORF y las regiones intergénicas del genoma de la levadura.[2]
Flujo de trabajo de un experimento de chip en chip
Un experimento ChIP-on-chip se puede dividir en tres pasos principales: el primero es configurar y diseñar el experimento seleccionando la matriz y el tipo de sonda apropiados. En segundo lugar, el experimento real se realiza en el laboratorio húmedo. Por último, durante la parte de laboratorio seco del ciclo, los datos recopilados se analizan para responder a la pregunta inicial o generar nuevas preguntas para que el ciclo pueda comenzar de nuevo.
Parte del flujo de trabajo Wet-lab
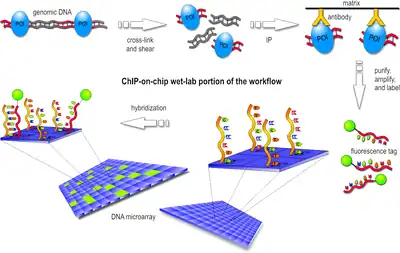
En el primer paso, la proteína de interés (POI) se entrecruza con el sitio de ADN al que se une en un entorno in vitro. Por lo general, esto se hace mediante una fijación suave con formaldehído que es reversible con calor.
Luego, las células se lisan y el ADN se corta mediante sonicación o usando nucleasa microcócica. Esto da como resultado fragmentos de ADN de doble cadena, normalmente de 1 kb o menos de longitud. Los que se entrecruzaron con el POI forman un complejo POI-ADN.
En el siguiente paso, solo estos complejos se filtran del conjunto de fragmentos de ADN, utilizando un anticuerpo específico para el POI. Los anticuerpos pueden estar unidos a una superficie sólida, pueden tener una perla magnética o alguna otra propiedad física que permita la separación de complejos entrecruzados y fragmentos no unidos. Este procedimiento es esencialmente una inmunoprecipitación (IP) de la proteína. Esto se puede hacer usando una proteína etiquetada con un anticuerpo contra la etiqueta (por ejemplo: FLAG, HA, c-myc) o con un anticuerpo contra la proteína nativa.
El entrecruzamiento de los complejos POI-DNA se invierte (normalmente mediante calentamiento) y las hebras de DNA se purifican. Para el resto del flujo de trabajo, el PDI ya no es necesario.
Después de un paso de amplificación y desnaturalización, los fragmentos de ADN monocatenario se etiquetan con una etiqueta fluorescente como Cy5 o Alexa 647.
Finalmente, los fragmentos se vierten sobre la superficie de la micromatriz de ADN, que se mancha con secuencias cortas monocatenarias que cubren la porción genómica de interés. Cada vez que un fragmento marcado "encuentra" un fragmento complementario en la matriz, se hibridarán y formarán nuevamente un fragmento de ADN de doble cadena.
Parte del flujo de trabajo Dry-lab
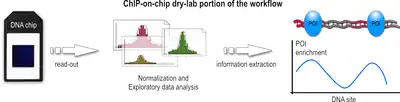
Después de un marco de tiempo suficientemente largo para permitir la hibridación, la matriz se ilumina con luz fluorescente. Esas sondas en la matriz que se hibridan con uno de los fragmentos etiquetados emiten una señal de luz que es capturada por una cámara. Esta imagen contiene todos los datos sin procesar de la parte restante del flujo de trabajo.
Estos datos sin procesar, codificados como una imagen de color falso, deben convertirse a valores numéricos antes de que se pueda realizar el análisis real. El análisis y la extracción de información de los datos sin procesar a menudo sigue siendo la parte más desafiante de los experimentos de chip en chip. Surgen problemas a lo largo de esta parte del flujo de trabajo, que van desde la lectura inicial del chip hasta los métodos adecuados para sustraer el ruido de fondo y, finalmente, los algoritmos apropiados que normalizan los datos y los ponen a disposición para el análisis estadístico posterior, que luego, con suerte, conducen a un una mejor comprensión de la cuestión biológica que el experimento busca abordar. Además, debido a las diferentes plataformas de arreglos y la falta de estandarización entre ellas, el almacenamiento e intercambio de datos es un gran problema. En términos generales, el análisis de datos se puede dividir en tres pasos principales:
Durante el primer paso, las señales de fluorescencia capturadas de la matriz se normalizan mediante señales de control derivadas del mismo chip o de un segundo chip. Estas señales de control indican qué sondas de la matriz se hibridaron correctamente y cuáles se unieron de forma no específica.
En el segundo paso, se aplican pruebas numéricas y estadísticas a los datos de control y los datos de fracciones de IP para identificar regiones enriquecidas con POI a lo largo del genoma. Los siguientes tres métodos se utilizan ampliamente: rango percentil mediano, error de matriz única y ventana deslizante. Estos métodos generalmente difieren en cómo se manejan las señales de baja intensidad, cuánto ruido de fondo se acepta y qué rasgo de los datos se enfatiza durante el cálculo. En el pasado reciente, el enfoque de ventana deslizante parece ser el favorito y, a menudo, se describe como el más poderoso.
En el tercer paso, estas regiones se analizan más a fondo. Si el POI fuera un factor de transcripción, tales regiones representarían sus sitios de unión. Luego, el análisis posterior puede querer inferir motivos de nucleótidos y otros patrones para permitir la anotación funcional del genoma.[4]
Fortalezas y debilidades
Usando matrices en mosaico, ChIP-on-chip permite una alta resolución de mapas de todo el genoma. Estos mapas pueden determinar los sitios de unión de muchas proteínas de unión al ADN, como factores de transcripción y también modificaciones de la cromatina.
Aunque ChIP-on-chip puede ser una técnica poderosa en el área de la genómica, es muy costosa. La mayoría de los estudios publicados que utilizan ChIP-on-chip repiten sus experimentos al menos tres veces para garantizar mapas biológicamente significativos. El costo de los microarrays de ADN es a menudo un factor limitante para decidir si un laboratorio debe proceder con un experimento de chip en chip. Otra limitación es el tamaño de los fragmentos de ADN que se pueden lograr. La mayoría de los protocolos ChIP-on-chip utilizan la sonicación como método para romper el ADN en pedazos pequeños. Sin embargo, la sonicación se limita a un tamaño de fragmento mínimo de 200 pb. Para mapas de mayor resolución, esta limitación debe superarse para lograr fragmentos más pequeños, preferiblemente a una resolución de un solo nucleosoma . Como se mencionó anteriormente, el análisis estadístico de la gran cantidad de datos generados a partir de arreglos es un desafío y los procedimientos de normalización deben apuntar a minimizar los artefactos y determinar qué es realmente significativo desde el punto de vista biológico. Hasta ahora, la aplicación a genomas de mamíferos ha sido una limitación importante, por ejemplo, debido al porcentaje significativo del genoma que está ocupado por repeticiones. Sin embargo, a medida que avanza la tecnología ChIP-on-chip, los mapas completos del genoma de los mamíferos de alta resolución deberían ser factibles.
Los anticuerpos utilizados para ChIP-on-chip pueden ser un factor limitante importante. ChIP-on-chip requiere anticuerpos altamente específicos que deben reconocer su epítopo en solución libre y también en condiciones fijas. Si se demuestra que inmunoprecipita con éxito la cromatina reticulada, se denomina "grado ChIP". Las empresas que proporcionan anticuerpos de grado ChIP incluyen Abcam, Cell Signaling Technology, Santa Cruz y Upstate. Para superar el problema de la especificidad, la proteína de interés se puede fusionar con una etiqueta como FLAG o HA que son reconocidas por los anticuerpos. Una alternativa a ChIP-on-chip que no requiere anticuerpos es DamID.
También están disponibles anticuerpos contra una modificación de histona específica como H3 tri metil K4. Como se mencionó anteriormente, la combinación de estos anticuerpos y ChIP-on-chip se ha vuelto extremadamente poderosa para determinar el análisis del genoma completo de los patrones de modificación de histonas y contribuirá enormemente a nuestra comprensión del código de histonas y la epigenética.
Un estudio que demuestra la naturaleza no específica de las proteínas de unión al ADN ha sido publicado en PLoS Biology. Esto indica que la confirmación alternativa de la relevancia funcional es un paso necesario en cualquier experimento de chip-chip.[5]
Historia
En 1999 se realizó un primer experimento de chip en chip para analizar la distribución de cohesina a lo largo del cromosoma III de levadura en ciernes.[6] Aunque el genoma no estaba completamente representado, el protocolo de este estudio sigue siendo equivalente a los utilizados en estudios posteriores. La técnica ChIP-on-chip que utiliza todos los ORF del genoma (que, sin embargo, permanece incompleto y le faltan regiones intergénicas) se aplicó con éxito en tres artículos publicados en 2000 y 2001.[7][8][9] Los autores identificaron sitios de unión para factores de transcripción individuales en la levadura en ciernes Saccharomyces cerevisiae. En 2002[10] se determinó las posiciones de todo el genoma de 106 factores de transcripción utilizando un sistema de etiquetado c-Myc en levadura. La primera demostración de la técnica ChIp-on-chip de mamíferos informó que el laboratorio de Peggy Farnham, en colaboración con el laboratorio de Michael Zhang, realizó el aislamiento de nueve fragmentos de cromatina que contenían un sitio de unión E2F débil y fuerte y se publicó en 2001.[11] Este estudio fue seguido varios meses después en una colaboración entre el laboratorio de Young y el laboratorio de Brian Dynlacht, que utilizó la técnica ChIP-on-chip para mostrar por primera vez que los objetivos E2F codifican componentes del punto de control del daño del ADN y las vías de reparación, como así como los factores involucrados en el ensamblaje/condensación de la cromatina, la segregación cromosómica y el punto de control del huso mitótico[12] Otras aplicaciones para ChIP-on-chip incluyen la replicación del ADN, la recombinación y la estructura de la cromatina. Desde entonces, ChIP-on-chip se ha convertido en una poderosa herramienta para determinar mapas de modificaciones de histonas de todo el genoma y muchos más factores de transcripción. ChIP-on-chip en sistemas de mamíferos ha sido difícil debido a los genomas grandes y repetitivos. Por lo tanto, muchos estudios en células de mamíferos se han centrado en regiones promotoras seleccionadas que se predice que se unen a factores de transcripción y no han analizado el genoma completo. Sin embargo, las matrices de genomas completos de mamíferos han estado disponibles comercialmente recientemente de compañías como Nimblegen. En el futuro, a medida que las matrices ChIP-on-chip se vuelvan cada vez más avanzadas, se analizarán con más detalle los mapas del genoma completo de alta resolución de las proteínas de unión al ADN y los componentes de la cromatina de los mamíferos.
Alternativas
- Introducida en 2007, la secuenciación ChIP (ChIP-seq) es una tecnología que utiliza la inmunoprecipitación de la cromatina para entrecruzar las proteínas de interés con el ADN, pero luego, en lugar de utilizar una micromatriz, utiliza el método de secuenciación más preciso y de mayor rendimiento para localizar puntos de interacción.[13]
- DamID es un método alternativo que no requiere anticuerpos.
- ChIP-exo utiliza el tratamiento con exonucleasa para lograr una resolución de hasta un solo par de bases.
- La secuenciación CUT&RUN utiliza el reconocimiento de anticuerpos con división enzimática dirigida para abordar algunas limitaciones técnicas de ChIP.
Referencias
- Aparicio, O; Geisberg, JV; Struhl, K (2004). «Chromatin immunoprecipitation for determining the association of proteins with specific genomic sequences in vivo». Current Protocols in Cell Biology. Chapter 17 (University of Southern California, Los Angeles, California, USA: John Wiley & Sons, Inc.). pp. Unit 17.7. ISBN 978-0-471-14303-1. ISSN 1934-2616. PMID 18228445. doi:10.1002/0471143030.cb1707s23.
- Buck, M. J.; Lieb, J. D. (2004). «ChIP-chip: Considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments». Genomics 83 (3): 349-360. PMID 14986705. doi:10.1016/j.ygeno.2003.11.004.
- Royce, T. E.; Rozowsky, J. S.; Bertone, P.; Samanta, M.; Stolc, V.; Weissman, S.; Snyder, M.; Gerstein, M. (2005). «Issues in the analysis of oligonucleotide tiling microarrays for transcript mapping». Trends in Genetics 21 (8): 466-475. PMC 1855044. PMID 15979196. doi:10.1016/j.tig.2005.06.007.
- «Biomedical Genomics Core - The Research Institute at Nationwide Children's Hospital». genomics.nchresearch.org.
- Li, Xiao-Yong; MacArthur, Stewart; Bourgon, Richard; Nix, David; Pollard, Daniel A.; Iyer, Venky N.; Hechmer, Aaron; Simirenko, Lisa et al. (2008). «Transcription Factors Bind Thousands of Active and Inactive Regions in the Drosophila Blastoderm». PLOS Biology 6 (2): e27. PMC 2235902. PMID 18271625. doi:10.1371/journal.pbio.0060027.
- Blat, Y.; Kleckner, N. (1999). «Cohesins bind to preferential sites along yeast chromosome III, with differential regulation along arms versus the centric region». Cell 98 (2): 249-259. PMID 10428036. doi:10.1016/s0092-8674(00)81019-3.
- Lieb, J. D.; Liu, X.; Botstein, D.; Brown, P. O. (2001). «Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association». Nature Genetics 28 (4): 327-334. PMID 11455386. doi:10.1038/ng569.
- Ren, B.; Robert, F.; Wyrick, J. J.; Aparicio, O.; Jennings, E. G.; Simon, I.; Zeitlinger, J.; Schreiber, J. et al. (2000). «Genome-wide location and function of DNA binding proteins». Science 290 (5500): 2306-2309. Bibcode:2000Sci...290.2306R. PMID 11125145. doi:10.1126/science.290.5500.2306.
- Iyer, V. R.; Horak, C. E.; Scafe, C. S.; Botstein, D.; Snyder, M.; Brown, P. O. (2001). «Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF». Nature 409 (6819): 533-538. Bibcode:2001Natur.409..533I. PMID 11206552. doi:10.1038/35054095.
- Lee, T. I.; Rinaldi, N. J.; Robert, F.; Odom, D. T.; Bar-Joseph, Z.; Gerber, G. K.; Hannett, N. M.; Harbison, C. T. et al. (2002). «Transcriptional regulatory networks in Saccharomyces cerevisiae». Science 298 (5594): 799-804. Bibcode:2002Sci...298..799L. PMID 12399584. doi:10.1126/science.1075090.
- Weinmann, A. S.; Bartley, S. M.; Zhang, T.; Zhang, M. Q.; Farnham, P. J. (2001). «Use of chromatin immunoprecipitation to clone novel E2F target promoters». Molecular and Cellular Biology 21 (20): 6820-6832. PMC 99859. PMID 11564866. doi:10.1128/MCB.21.20.6820-6832.2001.
- Ren, B.; Cam, H.; Takahashi, Y.; Volkert, T.; Terragni, J.; Young, R. A.; Dynlacht, B. D. (2002). «E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints». Genes & Development 16 (2): 245-256. PMC 155321. PMID 11799067. doi:10.1101/gad.949802.
- Johnson, David S.; Mortazavi, Ali; Myers, Richard M.; Wold, Barbara (8 de junio de 2007). «Genome-Wide Mapping of in Vivo Protein-DNA Interactions». Science 316 (5830): 1497-1502. Bibcode:2007Sci...316.1497J. PMID 17540862. doi:10.1126/science.1141319.
Otras lecturas
- Johnson, W. E.; Li, W.; Meyer, C. A.; Gottardo, R.; Carroll, J. S.; Brown, M.; Liu, X. S. (2006). «Model-based analysis of tiling-arrays for ChIP-chip». Proceedings of the National Academy of Sciences 103 (33): 12457-12462. Bibcode:2006PNAS..10312457J. ISSN 0027-8424. PMC 1567901. PMID 16895995. doi:10.1073/pnas.0601180103.
- Benoukraf, Touati; Cauchy, Pierre; Fenouil, Romain; Jeanniard, Adrien; Koch, Frederic; Jaeger, Sébastien; Thieffry, Denis; Imbert, Jean et al. (2009). «CoCAS: a ChIP-on-chip analysis suite». Bioinformatics 25 (7): 954-955. ISSN 1460-2059. PMC 2660873. PMID 19193731. doi:10.1093/bioinformatics/btp075.
Enlaces externos
- http://www.genome.gov/10005107 Proyecto ENCODE
- Información del paquete Chip-on-Chip (CoC) de Amkor Technology
Análisis y software
- CoCAS: un software de análisis gratuito para experimentos ChIP-on-Chip de Agilent
- rMAT: implementación de R del programa MAT para normalizar y analizar matrices de mosaico y datos de chip-chip.
| Control de autoridades |
|
|---|
 Datos: Q7815764
Datos: Q7815764