Ecología molecular
La ecología molecular es un campo de la biología evolutiva[1] que se ocupa de aplicar la genética de poblaciones moleculares, la filogenética molecular y, más recientemente, la genómica a cuestiones ecológicas tradicionales (diagnóstico de especies, conservación y evaluación de la biodiversidad, relaciones especies-área, etc. preguntas en ecología del comportamiento). Es virtualmente sinónimo del campo de la "Genética Ecológica", como lo iniciaron Theodosius Dobzhansky, E. B. Ford, Godfrey M. Hewitt y otros.[2] Estos campos están unidos en su intento de estudiar cuestiones de base genética "en el campo" en lugar de en el laboratorio. La ecología molecular está relacionada con el campo de la genética de la conservación.
Los métodos incluyen con frecuencia el uso de microsatélites para determinar el flujo de genes y la hibridación entre poblaciones. El desarrollo de la ecología molecular también está estrechamente relacionado con el uso de microarrays de ADN, lo que permite el análisis simultáneo de la expresión de miles de genes diferentes. La PCR cuantitativa también puede usarse para analizar la expresión génica como resultado de cambios en las condiciones ambientales o diferentes respuestas de individuos adaptados de manera diferente.
La ecología molecular utiliza datos genéticos moleculares para responder preguntas ecológicas relacionadas con biogeografía, genómica, genética de conservación y ecología del comportamiento. Los estudios utilizan principalmente datos basados en secuencias de ácido desoxirribonucleico (ADN). Este enfoque se ha mejorado durante varios años para permitir a los investigadores secuenciar miles de genes a partir de una pequeña cantidad de ADN inicial. Los tamaños de los alelos son otra forma en que los investigadores pueden comparar individuos y poblaciones, lo que les permite cuantificar la diversidad genética dentro de una población y las similitudes genéticas entre poblaciones.[3]

Diversidad bacteriana
Las técnicas de ecología molecular se utilizan para estudiar in situ cuestiones de diversidad bacteriana. Muchos microorganismos no se pueden obtener fácilmente como cepas cultivadas en el laboratorio, lo que permitiría su identificación y caracterización. También surge del desarrollo de la técnica de PCR, que permite la rápida amplificación del material genético.
La amplificación de ADN de muestras ambientales utilizando cebadores generales o específicos de grupo conduce a una mezcla de material genético, que requiere clasificación antes de secuenciar e identificar. La técnica clásica para lograrlo es mediante la clonación, que consiste en incorporar los fragmentos de ADN amplificados en plásmidos bacterianos. Técnicas como la electroforesis en gel de gradiente de temperatura permiten un resultado más rápido. Más recientemente, el advenimiento de tecnologías de secuenciación de ADN de próxima generación de costo relativamente bajo, como las plataformas 454 e Illumina, ha permitido la exploración de la ecología bacteriana con respecto a gradientes ambientales a escala continental, como el pH[4] que no era factible con la tecnología tradicional.

Diversidad de hongos
La exploración de la diversidad fúngica in situ también se ha beneficiado de las tecnologías de secuenciación de ADN de próxima generación. El uso de técnicas de secuenciación de alto rendimiento ha sido ampliamente adoptado por la comunidad de ecología fúngica desde la primera publicación de su uso en el campo en 2009.[5] De forma similar a la exploración de la diversidad bacteriana, estas técnicas han permitido estudios de alta resolución de cuestiones fundamentales en la ecología de los hongos como la filogeografía,[6] la diversidad de hongos en los suelos forestales,[7] estratificación de las comunidades de hongos en los horizontes del suelo,[8] y sucesión de hongos en la hojarasca vegetal en descomposición.[9]
La mayoría de las investigaciones sobre ecología fúngica que aprovechan los enfoques de secuenciación de próxima generación implican la secuenciación de amplicones de PCR de regiones conservadas de ADN (es decir, genes marcadores) para identificar y describir la distribución de grupos taxonómicos en la comunidad fúngica en cuestión, aunque la investigación más reciente se ha centrado en secuenciación de amplicones de genes funcionales.[5][8] El locus de elección para una descripción de la estructura taxonómica de las comunidades fúngicas ha sido tradicionalmente la región del espaciador transcrito interno (ITS) de los genes de ARN ribosómico[10] debido a su utilidad para identificar hongos a niveles taxonómicos de género o especie,[11] y su alta representación en bases de datos de secuencia pública. Un segundo locus ampliamente utilizado,[6][12] la región D1-D3 de los genes de ARN ribosómico 28S, puede no permitir la clasificación de bajo nivel taxonómico del ITS,[13][14] pero demuestra un rendimiento superior en la alineación de secuencias y la filogenética.[15] Además, la región D1-D3 puede ser un mejor candidato para la secuenciación con las tecnologías de secuenciación de Illumina.[16] Se mostró que la precisión de la clasificación de las secuencias de la región ITS o D1-D3 se basaba en gran medida en la composición de la secuencia y la calidad de las bases de datos utilizadas para la comparación, y las secuencias de mala calidad y la identificación errónea de secuencias en las bases de datos públicas es una preocupación importante.[17][18] La construcción de bases de datos de secuencias que tienen una amplia representación de los hongos y que son seleccionadas por expertos taxonómicos es el siguiente paso fundamental.[19]
Las tecnologías de secuenciación de próxima generación generan grandes cantidades de datos, y el análisis de datos de genes marcadores de hongos es un área activa de investigación.[5][20] Dos áreas principales de interés son los métodos para agrupar secuencias en unidades taxonómicas operativas mediante la similitud de secuencia y el control de calidad de los datos de secuencia. Actualmente, no hay consenso sobre los métodos preferidos para la agrupación, y la agrupación y los métodos de procesamiento de secuencias pueden afectar significativamente los resultados, especialmente para la región ITS de longitud variable. Además, las especies de hongos varían en la similitud de secuencia intraespecífica de la región ITS.[21] La investigación reciente se ha dedicado al desarrollo de protocolos de agrupación flexibles que permiten que los umbrales de similitud de secuencia varíen según los grupos taxonómicos, que están respaldados por secuencias bien anotadas en bases de datos de secuencias públicas.[19]
Fertilizaciones extrapares
En los últimos años, los datos y análisis moleculares han podido complementar los enfoques tradicionales de la ecología del comportamiento, el estudio del comportamiento animal en relación con su ecología y su historia evolutiva. Un comportamiento que los datos moleculares han ayudado a los científicos a comprender mejor son las fertilizaciones extrapares (EPF), también conocidas como copulaciones extrapares (EPC). Estos son eventos de apareamiento que ocurren fuera de un vínculo social, como la monogamia, y son difíciles de observar. Los datos moleculares han sido clave para comprender la prevalencia y las personas que participan en los EPF.
Si bien la mayoría de las especies de aves son socialmente monógamas, los datos moleculares han revelado que menos del 25% de estas especies son genéticamente monógamas.[22] Los EPF complican las cosas, especialmente para los hombres, porque no tiene sentido que un individuo cuide de la descendencia que no es suya. Los estudios han encontrado que los hombres ajustarán su cuidado parental en respuesta a cambios en su paternidad.[23][24] Otros estudios han demostrado que en especies socialmente monógamas, algunos individuos emplearán una estrategia alternativa para tener éxito reproductivo, ya que un vínculo social no siempre equivale al éxito reproductivo.[25][26]
Parece que los EPF en algunas especies son impulsados por la hipótesis de los genes buenos,[27] En alcaudones de lomo rojo (Lanius collurio), los machos extra-par tenían tarsos significativamente más largos que los machos dentro del par, y todos los extra-pares la descendencia eran machos, lo que apoya la predicción de que las hembras inclinarán su nidada hacia los machos cuando se aparean con un macho "atractivo".[28] En los reyezuelos domésticos (Troglodytes aedon), también se descubrió que las crías de parejas adicionales tenían un sesgo masculino en comparación con las crías internas.[29]
Sin la ecología molecular, sería imposible identificar a los individuos que participan en las EPF y la descendencia que resulta de las EPF.
Aislamiento por distancia
El aislamiento por distancia (IBD, del inglés Isolation by distance), como el aislamiento reproductivo, es el efecto de las barreras físicas a las poblaciones que limitan la migración y reducen el flujo de genes. Cuanto menor sea la distancia entre las poblaciones, más probabilidades hay de que los individuos se dispersen y aparezcan y, por lo tanto, aumentar el flujo de genes.[30] El uso de datos moleculares, específicamente las frecuencias alélicas de individuos entre poblaciones en relación con su distancia geográfica, ayuda a explicar conceptos como dispersión sesgada por sexo, especiación y genética del paisaje.
La prueba de Mantel es una evaluación que compara la distancia genética con la distancia geográfica y es más apropiada porque no supone que las comparaciones sean independientes entre sí.[3] Hay tres factores principales que influyen en las posibilidades de encontrar una correlación de la IBD, que incluyen el tamaño de la muestra, el metabolismo y los taxones.[31] Por ejemplo, según el metanálisis, los ectotermos tienen más probabilidades que los endotermos de presentar una mayor IBD.
Teoría de la metapoblación
La teoría de la metapoblación dicta que una metapoblación consiste en poblaciones espacialmente distintas que interactúan entre sí en algún nivel y pasan por un ciclo de extinciones y recolonizaciones (es decir, a través de la dispersión).[32] El modelo de metapoblación más común es el modelo de extinción-recolonización, que explica los sistemas en los que poblaciones espacialmente distintas experimentan cambios estocásticos en el tamaño de la población que pueden llevar a la extinción a nivel de población. Una vez que esto ha ocurrido, la dispersión de individuos de otras poblaciones emigrará y "rescatará" a la población en ese sitio. Otros modelos de metapoblación incluyen el modelo de fuente-sumidero (modelo isla-continente) donde una (o varias) grandes poblaciones centrales producen dispersiones a poblaciones satélites más pequeñas que tienen una tasa de crecimiento poblacional de menos de uno y no podrían persistir sin la afluencia de la población principal.
La estructura de la metapoblación y las repetidas extinciones y recolonizaciones pueden afectar significativamente la estructura genética de una población. La recolonización por algunos dispersores conduce a cuellos de botella de población que reducirán el tamaño efectivo de la población (Ne), acelerarán la deriva genética y agotarán la variación genética. Sin embargo, la dispersión entre poblaciones en la metapoblación puede revertir o detener estos procesos a largo plazo. Por lo tanto, para que las subpoblaciones individuales se mantengan saludables, deben tener un tamaño de población grande o tener una tasa relativamente alta de dispersión con otras subpoblaciones. La ecología molecular se centra en el uso de pruebas para determinar las tasas de dispersión entre poblaciones y puede usar relojes moleculares para determinar cuándo se produjeron cuellos de botella históricos. A medida que el hábitat se vuelve más fragmentado, la dispersión entre poblaciones será cada vez más rara. Por lo tanto, las subpoblaciones que históricamente pueden haber sido conservadas por una estructura de metapoblación pueden comenzar a disminuir. El uso de marcadores mitocondriales o nucleares para monitorear la dispersión, junto con los valores Fst de la población y la riqueza alélica, puede proporcionar información sobre qué tan bien se está desempeñando una población y cómo se desempeñará en el futuro.
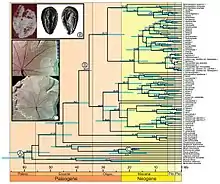
Hipótesis del reloj molecular
La hipótesis del reloj molecular establece que las secuencias de ADN evolucionan aproximadamente al mismo ritmo y, debido a esto, la disimilitud entre dos secuencias puede usarse para decir cuánto tiempo hace que divergieron entre sí. El primer paso para usar un reloj molecular es que debe calibrarse en función del tiempo aproximado en que los dos linajes estudiados divergieron. Las fuentes que se utilizan habitualmente para calibrar los relojes moleculares son fósiles o sucesos geológicos conocidos del pasado. Después de calibrar el reloj, el siguiente paso es calcular el tiempo de divergencia dividiendo el tiempo estimado desde que las secuencias divergieron por la cantidad de divergencia de secuencia. El número resultante es la tasa estimada a la que se está produciendo la evolución molecular. El reloj molecular más citado es un reloj de ADNmt "universal" de aproximadamente dos por ciento de divergencia de secuencia cada millón de años.[33] Aunque se conoce como un reloj universal, esta idea del reloj "universal" no es posible considerando que las tasas de evolución difieren dentro de las regiones del ADN. Otro inconveniente de usar relojes moleculares es que idealmente deben calibrarse a partir de una fuente de datos independiente distinta de los datos moleculares. Esto plantea un problema para los taxones que no se fosilizan/conservan fácilmente, lo que hace casi imposible calibrar su reloj molecular. A pesar de estos inconvenientes, la hipótesis del reloj molecular todavía se utiliza en la actualidad. El reloj molecular ha tenido éxito en fechar eventos que ocurrieron hasta hace 65 millones de años.[34]
%252C_mating.jpg.webp)
Hipótesis de elección de pareja
El concepto de elección de pareja explica cómo los organismos seleccionan a sus parejas basándose en dos métodos principales; la hipótesis de los buenos genes y la compatibilidad genética. La hipótesis de los buenos genes, también conocida como la hipótesis del hijo sexy, sugiere que las hembras elegirán un macho que produzca una descendencia que tendrá mayores ventajas de aptitud física y viabilidad genética. Por lo tanto, las parejas que son más 'atractivas' tienen más probabilidades de ser elegidas para aparearse y transmitir sus genes a la siguiente generación. En las especies que exhiben poliandria, las hembras buscarán los machos más adecuados y se volverán a aparear hasta que hayan encontrado el mejor esperma para fertilizar sus huevos.[35] La compatibilidad genética es donde los compañeros eligen a su pareja en función de la compatibilidad de sus genotipos. La pareja que realiza la selección debe conocer su propio genotipo, así como los genotipos de las parejas potenciales para poder seleccionar la pareja adecuada.[36] La compatibilidad genética en la mayoría de los casos se limita a rasgos específicos, como el principal complejo de histocompatibilidad en mamíferos, debido a complejas interacciones genéticas. Este comportamiento se ve potencialmente en humanos. Un estudio que analizó la elección de las mujeres en los hombres, basado en los olores corporales concluyó que el olor de los olores estaba influenciado por el MHC y que influían en la elección de pareja en las poblaciones humanas.[37]
Dispersión sesgada por sexo
La dispersión sesgada por sexo, o la tendencia de un sexo a dispersarse entre poblaciones con más frecuencia que el otro, es un comportamiento común estudiado por los investigadores. Actualmente existen tres hipótesis principales para ayudar a explicar la dispersión sesgada por sexo.[38] La hipótesis de la competencia por los recursos infiere que el sexo más filopátrico (el sexo con más probabilidades de permanecer en su suelo natal) se beneficia durante la reproducción simplemente por estar familiarizado con los recursos del suelo natal.[39] Una segunda propuesta para la dispersión sesgada por el sexo es la hipótesis de la competencia de pareja local, que introduce la idea de que los individuos encuentran menos competencia de pareja con sus parientes cuanto más se dispersan de sus lugares de origen.[40] Y la hipótesis de evitación de la endogamia sugiere que los individuos se dispersan para disminuir la endogamia.
Estudiar estas hipótesis puede ser arduo, ya que es casi imposible hacer un seguimiento de cada individuo y su paradero dentro y entre poblaciones. Para combatir este método que consume mucho tiempo, los científicos han reclutado varias técnicas de ecología molecular con el fin de estudiar la dispersión sesgada por sexo. Un método es la comparación de diferencias entre marcadores nucleares y mitocondriales entre poblaciones. Los marcadores que muestran niveles más altos de diferenciación indican el sexo más filopátrico; es decir, cuanto más permanezca un sexo en la tierra natal, más sus marcadores adquirirán una identificación única, debido a la falta de flujo genético con respecto a ese marcador.[41] Los investigadores también pueden cuantificar la relación de pareja hombre-hombre y mujer-mujer dentro de las poblaciones para comprender qué sexo tiene más probabilidades de dispersarse. Los pares con valores consistentemente más bajos en un sexo indican el sexo disperso. Esto se debe a que hay más flujo de genes en el sexo que se dispersa y sus marcadores son menos similares que los de los individuos del mismo sexo en la misma población, lo que produce un valor de parentesco bajo.[42] Los valores de FST también se utilizan para comprender los comportamientos de dispersión calculando un valor de FST para cada sexo. El sexo que se dispersa más muestra un valor de FST más bajo, que mide los niveles de consanguinidad entre la subpoblación y la población total. Además, las pruebas de asignación se pueden utilizar para cuantificar el número de individuos de un determinado sexo que se dispersan a otras poblaciones. Un enfoque más matemático para cuantificar la dispersión sesgada por sexo a nivel molecular es el uso de la autocorrelación espacial.[43] Esta correlación analiza la relación entre la distancia geográfica y la distancia espacial. Se calcula un coeficiente de correlación, o valor r, y la gráfica de r contra la distancia proporciona una indicación de los individuos más relacionados o menos relacionados entre sí de lo esperado.[27]
Loci de rasgos cuantitativos
Un locus de rasgo cuantitativo (QTL) se refiere a un conjunto de genes que controla un rasgo cuantitativo. Un rasgo cuantitativo es aquel que está influenciado por varios genes diferentes en lugar de uno o dos.[27] Los QTL se analizan usando Qst. El Qst analiza la relación de los rasgos en foco. En el caso de los QTL, las clinas se analizan mediante Qst. Una clina es un cambio en la frecuencia de los alelos a través de una distancia geográfica. Este cambio en la frecuencia de los alelos provoca una serie de fenotipos variables intermedios que cuando se asocian con ciertas condiciones ambientales pueden indicar selección. Esta selección provoca una adaptación local, pero todavía se espera que haya un alto flujo de genes a lo largo de la línea.
Por ejemplo, las lechuzas comunes en Europa exhiben una línea en referencia a la coloración de su plumaje. Sus plumas varían en coloración desde el blanco hasta el marrón rojizo en todo el rango geológico del suroeste al noreste.[44] Este estudio buscó encontrar si esta variación fenotípica se debía a la selección calculando los valores de Qst en las poblaciones de búhos. Debido a que todavía se anticipaba un alto flujo de genes a lo largo de esta línea, solo se esperaba que la selección actuara sobre los QTL que incurren en rasgos fenotípicos adaptativos localmente. Esto se puede determinar comparando los valores de Qst con los valores de FST (índice de fijación). Si ambos valores son similares y FST se basa en marcadores neutrales, entonces se puede suponer que los QTL también se basaron en marcadores neutrales (marcadores que no están bajo selección o adaptados localmente). Sin embargo, en el caso de las lechuzas comunes, el valor de Qst era mucho más alto que el valor de FST. Esto significa que estaba presente un alto flujo de genes, lo que permitía que los marcadores neutros fueran similares, indicado por el bajo valor de FST. Pero, la adaptación local debida a la selección también estuvo presente, en forma de coloración variable del plumaje, ya que el valor de Qst fue alto, lo que indica diferencias en estos loci no neutros. En otras palabras, esta línea de coloración del plumaje tiene algún tipo de valor adaptativo para las aves.
Índices de fijación
Los índices de fijación se utilizan para determinar el nivel de diferenciación genética entre subpoblaciones dentro de una población total. FST es el script que se usa para representar este índice cuando se usa la fórmula:

En esta ecuación, H T representa la heterocigosidad esperada de la población total y H S es la heterocigosidad esperada de una subpoblación. Ambas medidas de heterocigosidad se miden en un loci. En la ecuación, los valores de heterocigosidad esperados de la población total se comparan con los valores de heterocigosidad observados de las subpoblaciones dentro de esta población total. Los valores de FST más grandes implican que el nivel de diferenciación genética entre subpoblaciones dentro de una población total es más significativo.[27] El nivel de diferenciación es el resultado de un equilibrio entre el flujo de genes entre subpoblaciones (diferenciación decreciente) y la deriva genética dentro de estas subpoblaciones (diferenciación creciente); sin embargo, algunos ecologistas moleculares señalan que no se puede suponer que estos factores estén en equilibrio.[45] El FST también se puede ver como una forma de comparar la cantidad de endogamia dentro de las subpoblaciones con la cantidad de endogamia de la población total y, a veces, se lo denomina coeficiente de endogamia. En estos casos, los valores más altos de FST normalmente implican mayores cantidades de endogamia dentro de las subpoblaciones.[46] Otros factores, como las presiones de selección, también pueden afectar los valores de FST.[47]
Los valores de FST van acompañados de varias ecuaciones analógicas (FIS, GST, etc.). Estas medidas adicionales se interpretan de manera similar a los valores FST; sin embargo, se ajustan para acompañar a otros factores que FST puede que no, como la contabilidad de múltiples loci.[48]
_(14579196860).jpg.webp)
Depresión endogámica
La depresión endogámica es la reducción de la aptitud y la supervivencia de la descendencia de padres estrechamente relacionados.[49] La endogamia se observa comúnmente en poblaciones pequeñas debido a la mayor probabilidad de aparearse con un pariente debido a la limitada elección de pareja. Es más probable que la endogamia, especialmente en poblaciones pequeñas, resulte en tasas más altas de deriva genética, lo que conduce a tasas más altas de homocigosidad en todos los loci de la población y una menor heterocigosidad. La tasa de consanguinidad se basa en la disminución de la heterocigosidad. En otras palabras, la tasa a la que se pierde heterocigosidad de una población debido a la deriva genética es igual a la tasa de acumulación de endogamia en una población. En ausencia de migración, la endogamia se acumulará a una tasa inversamente proporcional al tamaño de la población.
Hay dos formas en que puede ocurrir la depresión endogámica. El primero de ellos es a través de la dominancia, donde los alelos beneficiosos suelen ser dominantes y los alelos dañinos suelen ser recesivos. El aumento de la homocigosidad resultante de la endogamia significa que es más probable que los alelos dañinos se expresen como homocigotos, y los efectos nocivos no pueden ser enmascarados por el alelo dominante beneficioso. El segundo método a través del cual se produce la depresión endogámica es a través de la sobredominancia o ventaja heterocigota.[50] Los individuos que son heterocigotos en un locus particular tienen una mayor aptitud que los homocigotos en ese locus. La endogamia conduce a una disminución de la heterocigosidad y, por lo tanto, a una disminución de la aptitud.
Los alelos deletéreos pueden eliminarse mediante selección natural de poblaciones consanguíneas mediante purga genética. A medida que aumenta la homocigosidad, se seleccionarán individuos menos aptos y, por lo tanto, esos alelos dañinos se perderán de la población.[27]
Depresión exógena
La depresión por exogamia es la reducción de la aptitud biológica en la descendencia de padres parientes lejanos.[51] La disminución de la aptitud debido a la exogamia se atribuye a una ruptura de complejos de genes coadaptados o relaciones epistáticas favorables. A diferencia de la depresión por endogamia, la depresión por endogamia enfatiza las interacciones entre los loci más que dentro de ellos. La depresión por endogamia y exogamia puede ocurrir al mismo tiempo. Los riesgos de depresión por exogamia aumentan con el aumento de la distancia entre poblaciones. El riesgo de exogamia durante el rescate genético a menudo limita la capacidad de aumentar la diversidad genética de un acervo genético pequeño o fragmentado. El engendro de un intermedio de dos o más rasgos adaptados puede hacer que la adaptación sea menos efectiva que cualquiera de las adaptaciones parentales.[52] Tres mecanismos principales influyen en la depresión exógena; deriva genética, cuellos de botella poblacionales, diferenciación de adaptaciones y diferencias cromosómicas establecidas que dan como resultado una descendencia estéril.[53] Si la exogamia es limitada y la población es lo suficientemente grande, la presión selectiva que actúa sobre cada generación puede restaurar la aptitud. Sin embargo, es probable que la población experimente un declive multigeneracional de la aptitud general, ya que la selección de rasgos lleva varias generaciones.[54] La selección actúa sobre las generaciones consanguíneas utilizando una mayor diversidad para adaptarse al medio ambiente. Esto puede resultar en una mayor aptitud entre la descendencia que el tipo parental original.
Unidades de conservación
Las unidades de conservación son clasificaciones que se utilizan a menudo en biología de la conservación, genética de la conservación y ecología molecular para separar y agrupar diferentes especies o poblaciones en función de la variación genética y la importancia para la protección.[55] Dos de los tipos más comunes de unidades de conservación son:
- Unidades de manejo (UM): Las unidades de manejo son poblaciones que tienen niveles muy bajos de flujo de genes y, por lo tanto, pueden diferenciarse genéticamente de otras poblaciones.[55]
- Unidades evolutivamente significativas (ESU): Las unidades evolutivamente significativas son poblaciones que muestran suficiente diferenciación genética para justificar su manejo como unidades distintas.
Las unidades de conservación a menudo se identifican utilizando marcadores genéticos neutrales y no neutrales, y cada uno tiene sus propias ventajas. El uso de marcadores neutrales durante la identificación de la unidad puede proporcionar suposiciones no sesgadas de la deriva genética y el tiempo transcurrido desde el aislamiento reproductivo dentro y entre especies y poblaciones, mientras que el uso de marcadores no neutrales puede proporcionar estimaciones más precisas de la divergencia evolutiva adaptativa, lo que puede ayudar a determinar el potencial de conservación para adaptarse a un determinado hábitat.[55]
Debido a las unidades de conservación, las poblaciones y especies que tienen niveles altos o diferentes de variación genética se pueden distinguir para manejar cada una individualmente, que en última instancia pueden diferir en función de una serie de factores. En un caso, el salmón del Atlántico ubicado dentro de la Bahía de Fundy recibió un significado evolutivo basado en las diferencias en las secuencias genéticas encontradas entre diferentes poblaciones.[56] Esta detección de importancia evolutiva puede permitir que cada población de salmón reciba conservación y protección personalizadas en función de su singularidad adaptativa en respuesta a la ubicación geográfica.

Filogenias y ecología comunitaria
Las filogenias son la historia evolutiva de un organismo, también conocida como filogeografía. Un árbol filogenético es un árbol que muestra relaciones evolutivas entre diferentes especies basadas en similitudes/diferencias entre rasgos genéticos o físicos. La ecología comunitaria se basa en el conocimiento de las relaciones evolutivas entre especies coexistentes.[57] Las filogenias abarcan aspectos tanto del tiempo (relaciones evolutivas) como del espacio (distribución geográfica).[58] Por lo general, los árboles de filogenia incluyen puntas, que representan grupos de especies descendientes, y nodos, que representan los antepasados comunes de esos descendientes. Si dos descendientes se separan del mismo nodo, se denominan grupos hermanos. También pueden incluir un grupo externo, una especie fuera del grupo de interés.[59] Los árboles representan clados, que es un grupo de organismos que incluye un antepasado y todos los descendientes de ese antepasado. El árbol de máxima parsimonia es el árbol más simple que tiene el número mínimo de pasos posible.
Las filogenias confieren importantes procesos históricos que dan forma a las distribuciones actuales de genes y especies.[58] Cuando dos especies se aíslan entre sí, retienen algunos de los mismos alelos ancestrales, también conocidos como alelos compartidos. Los alelos se pueden compartir debido a la clasificación e hibridación de linajes. La clasificación de linajes está impulsada por la deriva genética y debe ocurrir antes de que los alelos se vuelvan específicos de la especie. Algunos de estos alelos con el tiempo simplemente se perderán o pueden proliferar. La hibridación conduce a la introgresión de alelos de una especie a otra.[60]
La ecología comunitaria surgió de la historia natural y la biología de poblaciones. No solo incluye el estudio de las interacciones entre especies, sino que también se centra en conceptos ecológicos como el mutualismo, la depredación y la competencia dentro de las comunidades.[61] Se utiliza para explicar propiedades como la diversidad, el dominio y la composición de una comunidad.[62] Hay tres enfoques principales para integrar información filogenética en estudios de organizaciones comunitarias. El primer enfoque se centra en examinar la estructura filogenética de los ensamblajes comunitarios. El segundo enfoque se centra en explorar la base filogenética de las estructuras de nichos comunitarios. El camino final se centra en agregar un contexto comunitario a los estudios de evolución de rasgos y biogeografía.[57]
Conceptos de especies
Los conceptos de especies son objeto de debate en el campo de la ecología molecular. Desde el comienzo de la taxonomía, los científicos han querido estandarizar y perfeccionar la forma en que se definen las especies. Hay muchos conceptos de especies que dictan cómo los ecólogos determinan una buena especie. El concepto más comúnmente utilizado es el concepto de especie biológica que define una especie como grupos de poblaciones naturales real o potencialmente cruzadas, que están reproductivamente aisladas de otros grupos similares.[58] Este concepto no siempre es útil, especialmente cuando se trata de híbridos. Otros conceptos de especie incluyen el concepto de especie filogenética que describe una especie como el grupo monofilético identificable más pequeño de organismos dentro del cual existe un patrón parental de ascendencia y descendencia. Este concepto define especies en lo identificable. También sugeriría que hasta que dos grupos identificables realmente produzcan descendencia, seguirán siendo especies separadas. En 1999, John Avise y Glenn Johns sugirieron un método estandarizado para definir especies basado en especiaciones pasadas y medir clasificaciones biológicas como dependientes del tiempo. Su método utilizó bandas temporales para crear género, familia y orden en función de cuántas decenas de millones de años atrás tuvo lugar el evento de especiación que resultó en cada especie.[63]


Genética del paisaje
La genética del paisaje es un campo interdisciplinario que emerge rápidamente dentro de la ecología molecular. La genética del paisaje relaciona la genética con las características del paisaje, como el uso de la cobertura del suelo (bosques, agricultura, carreteras, etc.), la presencia de barreras y corredores, ríos, elevación, etc. La genética del paisaje responde cómo el paisaje afecta la dispersión y el flujo de genes.
Las barreras son cualquier característica del paisaje que evita la dispersión.[27] Las barreras para las especies terrestres pueden incluir montañas, ríos, carreteras y terrenos inadecuados, como campos agrícolas. Las barreras para las especies acuáticas pueden incluir islas o presas. Las barreras son específicas de la especie; por ejemplo, un río es una barrera para un ratón de campo, mientras que un halcón puede volar sobre un río. Los pasillos son áreas sobre las que es posible la dispersión. Los corredores son tramos de hábitat adecuado y también pueden ser construidos por el hombre, como pasos elevados sobre carreteras y escaleras para peces en presas.
Los datos geográficos utilizados para la genética del paisaje pueden incluir datos recopilados por radares en aviones, datos de satélites terrestres, datos marinos recopilados por NOAA, así como cualquier otro dato ecológico. En genética del paisaje, los investigadores a menudo utilizan diferentes análisis para intentar determinar la mejor manera de que una especie viaje del punto A al punto B. El análisis de la ruta de menor costo utiliza datos geográficos para determinar la ruta más eficiente de un punto a otro.[27] El análisis de escapes de circuitos predice todas las rutas posibles y la probabilidad de uso de cada ruta entre el punto A y el punto B. Estos análisis se utilizan para determinar la ruta que probablemente viajará un individuo en dispersión.
La genética del paisaje se está convirtiendo en una herramienta cada vez más importante en los esfuerzos de conservación de la vida silvestre. Se está utilizando para determinar cómo la pérdida y fragmentación del hábitat afecta el movimiento de especies.[64] También se utiliza para determinar qué especies deben gestionarse y si gestionar subpoblaciones de la misma forma o de forma diferente según su flujo de genes.
Véase también
Referencias
- Charlesworth, Brian. Measures of Divergence Between Populations and the Effect of Forces that Reduce Variability.
- Miglani (2015). Essentials of Molecular Genetics. Alpha Science International Limited. p. 36.
- Freeland, Joanna R. (2014). «Molecular Ecology». eLS. John Wiley & Sons, Inc. ISBN 978-0-470-01590-2. doi:10.1002/9780470015902.a0003268.pub2.
- Lauber, Christian L.; Hamady, Micah; Knight, Rob; Fierer, Noah (2009-08). «Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale». Applied and Environmental Microbiology 75 (15): 5111-5120. ISSN 1098-5336. PMC 2725504. PMID 19502440. doi:10.1128/AEM.00335-09.
- Větrovský, Tomáš; Baldrian, Petr (1 de noviembre de 2013). «Analysis of soil fungal communities by amplicon pyrosequencing: current approaches to data analysis and the introduction of the pipeline SEED». Biology and Fertility of Soils (en inglés) 49 (8): 1027-1037. ISSN 1432-0789. doi:10.1007/s00374-013-0801-y.
- Amend, Anthony S.; Seifert, Keith A.; Samson, Robert; Bruns, Thomas D. (3 de agosto de 2010). «Indoor fungal composition is geographically patterned and more diverse in temperate zones than in the tropics». Proceedings of the National Academy of Sciences of the United States of America 107 (31): 13748-13753. ISSN 1091-6490. PMC 2922287. PMID 20616017. doi:10.1073/pnas.1000454107.
- Buée, M.; Reich, M.; Murat, C.; Morin, E.; Nilsson, R. H.; Uroz, S.; Martin, F. (2009-10). «454 Pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity». The New Phytologist 184 (2): 449-456. ISSN 1469-8137. PMID 19703112. doi:10.1111/j.1469-8137.2009.03003.x.
- Baldrian, Petr; Kolařík, Miroslav; Štursová, Martina; Kopecký, Jan; Valášková, Vendula; Větrovský, Tomáš; Žifčáková, Lucia; Šnajdr, Jaroslav et al. (2012-02). «Active and total microbial communities in forest soil are largely different and highly stratified during decomposition». The ISME Journal 6 (2): 248-258. ISSN 1751-7362. PMC 3260513. PMID 21776033. doi:10.1038/ismej.2011.95.
- Voříšková, Jana; Baldrian, Petr (2013-03). «Fungal community on decomposing leaf litter undergoes rapid successional changes». The ISME Journal 7 (3): 477-486. ISSN 1751-7362. PMC 3578564. PMID 23051693. doi:10.1038/ismej.2012.116.
- Seifert, Keith A. (2009-05). «Progress towards DNA barcoding of fungi». Molecular Ecology Resources. 9 Suppl s1: 83-89. ISSN 1755-098X. PMID 21564968. doi:10.1111/j.1755-0998.2009.02635.x.
- Horton, T. R.; Bruns, T. D. (2001-08). «The molecular revolution in ectomycorrhizal ecology: peeking into the black-box». Molecular Ecology 10 (8): 1855-1871. ISSN 0962-1083. PMID 11555231. doi:10.1046/j.0962-1083.2001.01333.x.
- Weber, Carolyn F.; Vilgalys, Rytas; Kuske, Cheryl R. (9 de abril de 2013). «Changes in Fungal Community Composition in Response to Elevated Atmospheric CO2 and Nitrogen Fertilization Varies with Soil Horizon». Frontiers in Microbiology 4. ISSN 1664-302X. PMC 3621283. PMID 23641237. doi:10.3389/fmicb.2013.00078.
- Schoch, Conrad L.; Seifert, Keith A. (3 de julio de 2012). «Reply to Kiss: Internal transcribed spacer (ITS) remains the best candidate as a universal DNA barcode marker for Fungi despite imperfections». Proceedings of the National Academy of Sciences of the United States of America 109 (27): E1812. ISSN 0027-8424. PMC 3390861. doi:10.1073/pnas.1207508109.
- Porras-Alfaro, Andrea; Liu, Kuan-Liang; Kuske, Cheryl R.; Xie, Gary (2014-02). «From genus to phylum: large-subunit and internal transcribed spacer rRNA operon regions show similar classification accuracies influenced by database composition». Applied and Environmental Microbiology 80 (3): 829-840. ISSN 1098-5336. PMC 3911224. PMID 24242255. doi:10.1128/AEM.02894-13.
- Kõljalg, Urmas; Larsson, Karl-Henrik; Abarenkov, Kessy; Nilsson, R. Henrik; Alexander, Ian J.; Eberhardt, Ursula; Erland, Susanne; Høiland, Klaus et al. (2005-06). «UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi». The New Phytologist 166 (3): 1063-1068. ISSN 0028-646X. PMID 15869663. doi:10.1111/j.1469-8137.2005.01376.x.
- Liu, Kuan-Liang; Porras-Alfaro, Andrea; Kuske, Cheryl R.; Eichorst, Stephanie A.; Xie, Gary (2012-03). «Accurate, rapid taxonomic classification of fungal large-subunit rRNA genes». Applied and Environmental Microbiology 78 (5): 1523-1533. ISSN 1098-5336. PMC 3294464. PMID 22194300. doi:10.1128/AEM.06826-11.
- Vilgalys, Rytas (2003). «Taxonomic misidentification in public DNA databases». New Phytologist (en inglés) 160 (1): 4-5. ISSN 1469-8137. doi:10.1046/j.1469-8137.2003.00894.x.
- Nilsson, R. Henrik; Tedersoo, Leho; Abarenkov, Kessy; Ryberg, Martin; Kristiansson, Erik; Hartmann, Martin; Schoch, Conrad L.; Nylander, Johan A. A. et al. (9 de mayo de 2012). «Five simple guidelines for establishing basic authenticity and reliability of newly generated fungal ITS sequences». MycoKeys (en inglés) 4: 37-63. ISSN 1314-4049. doi:10.3897/mycokeys.4.3606.
- Kõljalg, Urmas; Nilsson, R. Henrik; Abarenkov, Kessy; Tedersoo, Leho; Taylor, Andy F. S.; Bahram, Mohammad; Bates, Scott T.; Bruns, Thomas D. et al. (2013-11). «Towards a unified paradigm for sequence-based identification of fungi». Molecular Ecology 22 (21): 5271-5277. ISSN 1365-294X. PMID 24112409. doi:10.1111/mec.12481.
- Lindahl, Björn D; Nilsson, R Henrik; Tedersoo, Leho; Abarenkov, Kessy; Carlsen, Tor; Kjøller, Rasmus; Kõljalg, Urmas; Pennanen, Taina et al. (2013-7). «Fungal community analysis by high-throughput sequencing of amplified markers – a user's guide». The New Phytologist 199 (1): 288-299. ISSN 0028-646X. PMC 3712477. PMID 23534863. doi:10.1111/nph.12243.
- Nilsson, R. Henrik; Kristiansson, Erik; Ryberg, Martin; Hallenberg, Nils; Larsson, Karl-Henrik (26 de mayo de 2008). «Intraspecific ITS variability in the kingdom fungi as expressed in the international sequence databases and its implications for molecular species identification». Evolutionary Bioinformatics Online 4: 193-201. ISSN 1176-9343. PMC 2614188. PMID 19204817. doi:10.4137/ebo.s653.
- Griffith, Simon C.; Owens, Ian P. F.; Thuman, Katherine A. (2002-11). «Extra pair paternity in birds: a review of interspecific variation and adaptive function». Molecular Ecology 11 (11): 2195-2212. ISSN 0962-1083. PMID 12406233. doi:10.1046/j.1365-294x.2002.01613.x.
- Hunt, J.; Simmons, L. W. (2002). «Confidence of paternity and paternal care: covariation revealed through the experimental manipulation of the mating system in the beetle Onthophagus taurus». Journal of Evolutionary Biology (en inglés) 15 (5): 784-795. ISSN 1420-9101. doi:10.1046/j.1420-9101.2002.00442.x.
- Neff, Bryan D. (17 de abril de 2003). «Decisions about parental care in response to perceived paternity». Nature 422 (6933): 716-719. ISSN 0028-0836. PMID 12700761. doi:10.1038/nature01528.
- Conrad, K. F.; Johnston, P. V.; Crossman, C.; Kempenaers, B.; Robertson, R. J.; Wheelwright, N. T.; Boag, P. T. (2001-05). «High levels of extra-pair paternity in an isolated, low-density, island population of tree swallows (Tachycineta bicolor)». Molecular Ecology 10 (5): 1301-1308. ISSN 0962-1083. PMID 11380885. doi:10.1046/j.1365-294x.2001.01263.x.
- Kempenaers, Bart; Everding, Susie; Bishop, Cheryl; Boag, Peter; Robertson, Raleigh J. (1 de marzo de 2001). «Extra-pair paternity and the reproductive role of male floaters in the tree swallow (Tachycineta bicolor)». Behavioral Ecology and Sociobiology (en inglés) 49 (4): 251-259. ISSN 1432-0762. doi:10.1007/s002650000305.
- Freeland, Joanna; Kirk, Heather; Petersen, Stephen (2011). Molecular Ecology. West Sussex, UK: John Wiley & Sons, Ltd. p. 295. ISBN 978-1-119-99308-7.
- Schwarzová, Lucie; Šimek, Jaroslav; Coppack, Timothy; Tryjanowski, Piotr (1 de junio de 2008). «Male-Biased Sex of Extra Pair Young in the Socially Monogamous Red-Backed Shrike Lanius collurio». Acta Ornithologica 43 (2): 235-239. ISSN 0001-6454. doi:10.3161/000164508X395379.
- Johnson, L. Scott; Thompson, Charles F.; Sakaluk, Scott K.; Neuhäuser, Markus; Johnson, Bonnie G.P.; Soukup, Sheryl Swartz; Forsythe, Shannon Janota; Masters, Brian S. (22 de junio de 2009). «Extra-pair young in house wren broods are more likely to be male than female». Proceedings of the Royal Society B: Biological Sciences 276 (1665): 2285-2289. ISSN 0962-8452. PMC 2677618. PMID 19324727. doi:10.1098/rspb.2009.0283.
- Wright, S. (1943-03). «Isolation by Distance». Genetics 28 (2): 114-138. ISSN 0016-6731. PMC 1209196. PMID 17247074.
- Jenkins, David G.; Carey, Michael; Czerniewska, Justyna; Fletcher, Jennifer; Hether, Tyler; Jones, Amanda; Knight, Stacy; Knox, Joseph et al. (2010). «A meta-analysis of isolation by distance: relic or reference standard for landscape genetics?». Ecography (en inglés) 33 (2): 315-320. ISSN 1600-0587. doi:10.1111/j.1600-0587.2010.06285.x.
- Levins, R. (1 de septiembre de 1969). «Some Demographic and Genetic Consequences of Environmental Heterogeneity for Biological Control». Bulletin of the Entomological Society of America 15 (3): 237-240. ISSN 0013-8754. doi:10.1093/besa/15.3.237.
- Brown, W. M.; Prager, E. M.; Wang, A.; Wilson, A. C. (1982). «Mitochondrial DNA sequences of primates: tempo and mode of evolution». Journal of Molecular Evolution 18 (4): 225-239. ISSN 0022-2844. PMID 6284948. doi:10.1007/BF01734101.
- Kumar, S.; Hedges, S. B. (30 de abril de 1998). «A molecular timescale for vertebrate evolution». Nature 392 (6679): 917-920. ISSN 0028-0836. PMID 9582070. doi:10.1038/31927.
- Yasui, Yukio (March 1997). «A "Good-Sperm" model can explain the evolution of costly multiple mating by females». The American Naturalist 149 (3): 573-584. doi:10.1086/286006.
- Puurtinen, Mikael; Ketola, Tarmo; Kotiaho, Janne S. (2005-04). «Genetic compatibility and sexual selection». Trends in Ecology & Evolution 20 (4): 157-158. ISSN 0169-5347. PMID 16701361. doi:10.1016/j.tree.2005.02.005.
- Wedekind, C.; Seebeck, T.; Bettens, F.; Paepke, A. J. (22 de junio de 1995). «MHC-dependent mate preferences in humans». Proceedings. Biological Sciences 260 (1359): 245-249. ISSN 0962-8452. PMID 7630893. doi:10.1098/rspb.1995.0087.
- Végvári, Zsolt; Katona, Gergely; Vági, Balázs; Freckleton, Robert P.; Gaillard, Jean‐Michel; Székely, Tamás; Liker, András (21 de junio de 2018). «Sex‐biased breeding dispersal is predicted by social environment in birds». Ecology and Evolution 8 (13): 6483-6491. ISSN 2045-7758. PMC 6053579. PMID 30038750. doi:10.1002/ece3.4095.
- Hjernquist, Mårten B.; Thuman Hjernquist, Katherine A.; Forsman, Jukka T.; Gustafsson, Lars (1 de marzo de 2009). «Sex allocation in response to local resource competition over breeding territories». Behavioral Ecology (en inglés) 20 (2): 335-339. ISSN 1045-2249. doi:10.1093/beheco/arp002.
- Herre, E. A. (17 de mayo de 1985). «Sex ratio adjustment in fig wasps». Science (New York, N.Y.) 228 (4701): 896-898. ISSN 0036-8075. PMID 17815055. doi:10.1126/science.228.4701.896.
- Verkuil, Yvonne I; Juillet, Cedric; Lank, David B; Widemo, Fredrik; Piersma, Theunis (2014-9). «Genetic variation in nuclear and mitochondrial markers supports a large sex difference in lifetime reproductive skew in a lekking species». Ecology and Evolution 4 (18): 3626-3632. ISSN 2045-7758. PMC 4224536. PMID 25478153. doi:10.1002/ece3.1188.
- Zhang, Liangzhi; Qu, Jiapeng; Li, Kexin; Li, Wenjing; Yang, Min; Zhang, Yanming (22 de agosto de 2017). «Genetic diversity and sex‐bias dispersal of plateau pika in Tibetan plateau». Ecology and Evolution 7 (19): 7708-7718. ISSN 2045-7758. PMC 5632614. PMID 29043027. doi:10.1002/ece3.3289.
- «Spatial autocorrelation — R Spatial». rspatial.org. Consultado el 3 de abril de 2020.
- Antoniazza, Sylvain; Burri, Reto; Fumagalli, Luca; Goudet, Jérôme; Roulin, Alexandre (2010-07). «Local adaptation maintains clinal variation in melanin-based coloration of European barn owls (Tyto alba)». Evolution; International Journal of Organic Evolution 64 (7): 1944-1954. ISSN 1558-5646. PMID 20148951. doi:10.1111/j.1558-5646.2010.00969.x.
- Woiwood, Ian; Reynolds, D. R.; Thomas, C. D. (2001). Insect Movement: Mechanisms and Consequences : Proceedings of the Royal Entomological Society's 20th Symposium (en inglés). CABI. ISBN 978-0-85199-781-0.
- «Inbreeding and Population Structure». University of Vermont.
- Hu, Xin-Sheng; He, Fangliang (21 de julio de 2005). «Background selection and population differentiation». Journal of Theoretical Biology 235 (2): 207-219. ISSN 0022-5193. PMID 15862590. doi:10.1016/j.jtbi.2005.01.004.
- «An Introduction to Restoration Genetics: How is Genetic Diversity Distributed in Natural Populations?». www.nps.gov.
- Charlesworth, Deborah; Willis, John H. (2009-11). «The genetics of inbreeding depression». Nature Reviews. Genetics 10 (11): 783-796. ISSN 1471-0064. PMID 19834483. doi:10.1038/nrg2664.
- Charlesworth, Deborah; Willis, John H. (November 2009). «The genetics of inbreeding depression». Nature Reviews Genetics (en inglés) 10 (11): 783-796. ISSN 1471-0064. PMID 19834483. doi:10.1038/nrg2664.
- Frankham, Richard; Ballou, Jonathan D.; Eldridge, Mark D. B.; Lacy, Robert C.; Ralls, Katherine; Dudash, Michele R.; Fenster, Charles B. (2011-06). «Predicting the probability of outbreeding depression». Conservation Biology: The Journal of the Society for Conservation Biology 25 (3): 465-475. ISSN 1523-1739. PMID 21486369. doi:10.1111/j.1523-1739.2011.01662.x.
- Turcek, F. J.; Hickey, J. J. (January 1951). «Effect of Introductions on Two Game Populations in Czechoslovakia». The Journal of Wildlife Management 15 (1): 113. doi:10.2307/3796784.
- Fenster, Charles B.; Galloway, Laura F. (18 de octubre de 2000). «Inbreeding and Outbreeding Depression in Natural Populations of Chamaecrista fasciculata (Fabaceae)». Conservation Biology (en inglés) 14 (5): 1406-1412. ISSN 0888-8892. doi:10.1046/j.1523-1739.2000.99234.x.
- Frankham, Richard; Ballou, Jonathan D; Briscoe, David A; McInness, Karina H (2010). Introduction to conservation genetics (2nd edición). Cambridge, UK: Cambridge University Press. ISBN 978-0-521-87847-0. OCLC 268793768.
- Freeland, Joanna; Kirk, Heather; Petersen, Stephen (2011). Molecular Ecology. Chichester, West Sussex, UK: John Wiley & Sons. pp. 332-333. ISBN 978-0-470-74833-6.
- Vandersteen Tymchuk, Wendy; O'Reilly, Patrick; Bittman, Jesse; Macdonald, Danielle; Schulte, Patricia (2010-05). «Conservation genomics of Atlantic salmon: variation in gene expression between and within regions of the Bay of Fundy». Molecular Ecology 19 (9): 1842-1859. ISSN 1365-294X. PMID 20529070. doi:10.1111/j.1365-294X.2010.04596.x.
- Webb, Campbell O.; Ackerly, David D.; McPeek, Mark A.; Donoghue, Michael J . (November 2002). «Phylogenies and Community Ecology». Annual Review of Ecology and Systematics (en inglés) 33 (1): 475-505. ISSN 0066-4162. doi:10.1146/annurev.ecolsys.33.010802.150448.
- Freeland, Joanna; Petersen, Stephen; Kirk, Heather (2011). Molecular Ecology (2nd edición). Chichester, West Sussex, UK: Wiley-Blackwell.
- «Reading trees: A quick review». Understanding Evolution. (University of California Museum of Paleontology.). 22 de agosto de 2008.
- Harrison, Larson, Richard, Erica (2014). «Hybridization, introgression, and the nature of species boundaries». Journal of Heredity 105.
- «Community ecology - Latest research and news | Nature». www.nature.com. Consultado el 7 de marzo de 2020.
- Jackson, Stephen T.; Blois, Jessica L. (21 de abril de 2015). «Community ecology in a changing environment: Perspectives from the Quaternary». Proceedings of the National Academy of Sciences of the United States of America 112 (16): 4915-4921. ISSN 0027-8424. PMC 4413336. PMID 25901314. doi:10.1073/pnas.1403664111.
- Avise, John C.; Johns, Glenn C. (22 de junio de 1999). «Proposal for a standardized temporal scheme of biological classification for extant species». Proceedings of the National Academy of Sciences of the United States of America 96 (13): 7358-7363. ISSN 0027-8424. PMID 10377419.
- Holderegger, Rolf; Wagner, Helene H. (1 de marzo de 2008). «Landscape Genetics». BioScience (en inglés) 58 (3): 199-207. doi:10.1641/B580306.
Enlaces externos
| Control de autoridades |
|
|---|
 Datos: Q1785846
Datos: Q1785846