Enfermedad de Creutzfeldt-Jakob
La encefalopatía espongiforme subaguda, mejor conocida como enfermedad de Creutzfeldt-Jakob (ECJ) es un trastorno neurológico poco frecuente de carácter neurodegenerativo pero invariablemente fatal que evoluciona velozmente a la demencia y por último a la muerte, ocasionado por un plegamiento anómalo en una proteína llamada prion (PrP), que resulta en una mutación patogénica de su conformación tridimensional, habitualmente identificada como PrPSc. Los síntomas iniciales pueden incluir problemas de marcha, pérdida de la memoria, cambios en la personalidad, visión borrosa o ceguera, dificultad para pensar, insomnio, psicosis y movimientos bruscos e involuntarios (mioclonía). En un 10% de los casos sigue un patrón de herencia autosómico dominante, que predispone al afectado a la mutación patogénica del prion y en un 85-90% de los casos en donde la causa de la mutación del prion es desconocida el surgimiento de la enfermedad es esporádico. Se trata de una enfermedad de rápida progresión sintomatológica, con un pronóstico de sobrevivencia pobre o, generalmente, fatal que afecta aproximadamente a una persona por millón (prevalencia de 1:106) a nivel global.
| Enfermedad de Creutzfeldt-Jakob | ||
|---|---|---|
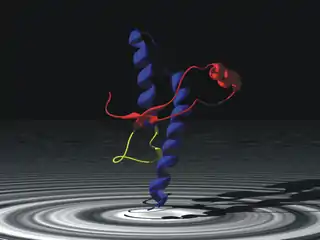 Modelo molecular del prion humano PrPC. | ||
| Especialidad |
neurología infectología | |
| Síntomas |
Tempranos: problemas de memoria, cambios de comportamiento, mala coordinación, alteraciones visuales [4] Posteriormente : demencia, movimientos involuntarios, ceguera, debilidad, coma | |
| Tipos | Esporádico (mutación), familiar (hereditario), iatrogénico (adquirido), variante ( infección) | |
De acuerdo con la evidencia disponible, la ECJ resulta del plegamiento anormal de un prion. Este fenómeno parece estimular a que otras proteínas alteren sus formas, afectando su capacidad para funcionar. Por esto, se la clasifica entre las enfermedades priónicas o encefalopatías espongiformes transmisibles (EET), caracterizadas por presentar una forma anómala de la proteína priónica celular (PrPC). Estas enfermedades pueden existir en formas esporádicas (idiopáticas), hereditarias, y adquiridas. El término espongiforme alude al aspecto esponjoso que presenta en la autopsia el cerebro afectado.[1]
Historia
Aunque es posible que la enfermedad se conociera desde la más remota antigüedad, sus síntomas inespecíficos deben de haber sido confundidos con otros tipos de demencia durante siglos. En 1920, el neuropatólogo alemán Alfons Maria Jakob describió una serie de casos de seis pacientes con espasticidad y demencia progresiva asociada a una degeneración neurológica. Poco tiempo más tarde, otro neuropatólogo alemán, Hans-Gerhard Creutzfeldt publicó, de manera independiente, un caso similar. Jakob atribuyó el descubrimiento de la enfermedad a Creutzfeldt por haber descrito el síndrome por primera vez, sin darse cuenta de que él mismo lo había hecho de forma paralela. El primero en hacer uso del término «enfermedad de Creutzfeldt-Jakob» fue Walther Spielmeyer, en el año 1922.[2] Algunos de los hallazgos clínicos que ellos describieron en sus primeros documentos sobre la ECJ no se corresponden con los criterios actuales sobre la misma, por lo que se considera altamente probable que algunos de los casos estudiados en sus investigaciones iniciales fueran víctimas de otra enfermedad.
La variante iatrogénica —es decir, de origen en el medio hospitalario— fue descrita por primera vez en 1974 en un paciente que había recibido un trasplante de córnea de un donante que sufría la variante espontánea de la enfermedad.[3]
En 1966 la ECJ tipo Kuru fue transmitida al chimpancé probando su capacidad infecciosa. Este carácter infeccioso de la enfermedad fue descrita luego en 1968.[4] Inicialmente se pensó que el agente causal era un virus, encontrándose luego que era una glucoproteína mal doblada y que puede tener condicionamiento genético. La proteína prion fue identificada en estudios por Stanley B. Prusiner en 1982 por lo que recibió el premio nobel de medicina en 1997.[5] Fue descubierto luego que el componente genético se encuentra en el brazo corto del cromosoma 20.
Epidemiología
El ser humano es el único reservorio conocido para el prion de la enfermedad de Creutzfeldt-Jakob (ECJ).[6] La ECJ aparece generalmente en la edad madura y evoluciona con rapidez, afectando en proporciones comparables a hombres y mujeres.[7] Globalmente, afecta a una o dos personas por millón anualmente.[8] Los síntomas comienzan a una edad media de 61 años y, el un 70 % de los pacientes fallece al cabo de un año.[9]
- Para la forma esporádica de la enfermedad específicamente (ECJe) —que corresponde a un 85 % del total de casos— la media de diagnóstico son los 67 años, con una supervivencia a menudo limitada a los cuatro meses. Esta media tan avanzada implica, sin embargo, que exista la posibilidad de que la patología se infradiagnostique al compartir sintomatología con otras enfermedades con alta incidencia en esos rangos de edad.[8]
- Para la variante causada por la encefalopatía espongiforme bovina (ECJv), la media de diagnóstico son los 26 años para aquellos que demuestran síntomas, y la supervivencia media es de 13-14 meses.[10] Entre el año 2000 y el 2020, su incidencia se ha reducido considerablemente.[8]
- En cuanto a la variante iatrogénica (ECJi), se hallaron por encima de 450 casos totales a escala global desde su descubrimiento en 1974 hasta 2017.[3]
La presencia de casos de la enfermedad suele correlacionarse con un aumento de la vigilancia de la misma, especialmente en países en los que se han dado epidemias de ECJ.[11] Otros factores que pueden contribuir a un registro mayor de incidencia en una sociedad son una edad media más avanzada, un aumento de la población, una mayor concienciación al respecto por parte de los profesionales de salud, y la existencia de métodos de diagnóstico más precisos. Ya que sus síntomas son similares a los de otras patologías neurológicas, existe la posibilidad de que la ECJ se confunda con ictus, nefropatía aguda, demencia general e hiperparatiroidismo en el diagnóstico.[11]
Cuadro clínico
Los primeros síntomas de la enfermedad de Creutzfeldt-Jakob incluyen típicamente demencia —cambios de personalidad junto con deterioro de la memoria, el juicio y el pensamiento— y problemas de coordinación muscular.[12] Las personas con la enfermedad también pueden experimentar insomnio, depresión o sensaciones inusitadas. La ECJ no ocasiona fiebre ni otros síntomas comunes. A medida que progresa la enfermedad, el deterioro mental del paciente se agudiza y aparecen síntomas piramidales y extrapiramidales como rigidez. A menudo comienza a tener contracciones musculares involuntarias llamadas mioclono y puede quedar ciego por defecto cerebral, perder el control de los esfínteres o una amplia variedad de otros síntomas neurológicos que amenazan con la comodidad y la vida de la persona. Con el tiempo se deteriora la capacidad de moverse y hablar y caen en coma. La neumonía y otras infecciones complican a menudo el curso de la enfermedad y pueden conducir a la muerte por sí mismas.
La nueva variante
Alrededor de 1920 se describieron algunos casos que probablemente fueran en realidad de ECJ adquirida, aunque muchos investigadores opinan que se trataba de otra enfermedad priónica denominada kuru, que se transmitía entre los nativos de la etnia fore de Nueva Guinea debido a los ritos funerarios de canibalismo familiar. El reservorio de la variante de la ECJ parece ser el ganado infectado por el prion de la encefalopatía espongiforme bovina.[6]
En 1996, se reportó en el Reino Unido la "nueva variante" de ECJ (ECJv),[nota 1] y se describieron diez casos ocurridos entre 1994 y 1995.[13] La nueva variante se puede transmitir por contagio entre distintas especies y, posiblemente, de persona a persona.[12] Actualmente se postula que, en algunos casos, la forma adquirida de la ECJ sería la patología humana subsiguiente a la infección con el llamado "mal de la vaca loca", nombre con que se conoce la encefalopatía espongiforme bovina (EEB). No obstante, cabe aclarar que en el último caso, tanto la leche, como el músculo, el tejido adiposo y los fluidos (saliva, sangre, orina, semen) del ganado bovino carecen de capacidad infectiva por vía oral.[14]
Desde 1995 y hasta la mitad del año 2008 se han registrado 204 casos de este tipo,[15] ocurridos mayoritariamente en Gran Bretaña.
Diagnóstico
El diagnóstico correcto de la ECJ es muy difícil, porque a menudo los síntomas pueden confundirse con los de otros trastornos neurológicos progresivos tales como el Alzheimer o la enfermedad de Huntington. Sin embargo, la ECJ ocasiona inconfundibles cambios en el tejido cerebral, claramente visibles en la autopsia. También tiende a ocasionar un deterioro más rápido de las capacidades del paciente que la enfermedad de Alzheimer o la mayoría de los demás tipos de demencia.
En la actualidad no hay una prueba diagnóstica certera para la enfermedad de Creutzfeldt-Jakob. Cuando un médico sospecha la presencia de ECJ, la primera preocupación consiste en descartar otras formas tratables de demencia tales como la encefalitis (inflamación del cerebro) o la meningitis crónica, por lo que se requiere la evaluación por un neurólogo cualificado. Las pruebas estándar de diagnóstico incluyen una punción espinal para descartar otras causas de demencia y un electroencefalograma (EEG) para registrar el patrón eléctrico del cerebro, que puede ser particularmente valioso ya que muestra un tipo específico de anomalía en la ECJ.[7]
La tomografía computarizada de cerebro puede ayudar a descartar la posibilidad de que los síntomas sean el resultado de otros problemas tales como un ataque al corazón o un tumor cerebral. Las exploraciones del cerebro mediante imágenes de resonancia magnética nuclear (RMN) también pueden poner de relieve patrones característicos de degeneración cerebral que ayuden a diagnosticar la ECJ.
La única forma de confirmar un diagnóstico de la ECJ es mediante una biopsia o autopsia cerebral. En una biopsia cerebral, el neurocirujano separa un pequeño trozo de tejido del cerebro del paciente a fin de que pueda examinarlo un neuropatólogo. Este procedimiento puede ser peligroso para el paciente y la operación no siempre obtiene el tejido de la parte afectada del cerebro. Debido a que un diagnóstico correcto de la ECJ no mejora el pronóstico ni las posibilidades de tratamiento, la biopsia cerebral no se aconseja a menos que se necesite para descartar un trastorno tratable. En una autopsia, se examina todo el cerebro después de la muerte.
Clasificación
Hay tres clases principales de la enfermedad de Creutzfeldt-Jakob (ECJ):
ECJ esporádica
En estos casos, la enfermedad se presenta aun cuando la persona parece estar libre de factores de riesgo asociados, es decir, la etiología es desconocida. Su alcance es mundial, siendo ocasionada a veces por una mutación sin sentido del gen de la proteína priónica (PRNP). Otras veces, el envejecimiento es el único factor de riesgo consistente.[14] Asimismo, se han identificado otras mutaciones que no causan directamente la enfermedad, pero vuelven a los individuos más susceptibles de contraer la infección con el prion. Estas últimas mutaciones estarían implicadas parcialmente en la incidencia esporádica de la enfermedad.
Este es el tipo más común de ECJ, manifestándose en, al menos, un 85 % de los casos. Sin embargo, no es posible adscribir directamente los casos de ECJ esporádica a los otros dos grupos. La revisión de los hallazgos clínicos en casos de ECJv reveló que estos diferían sustancialmente de los tradicionalmente observados en casos esporádicos.[16]
ECJ hereditaria

Se puede determinar en la historia del paciente algún antecedente familiar de la enfermedad o pruebas positivas de mutación genética asociada con el gen productor del prion causante de la ECJ. En los Estados Unidos, entre el 5 y el 10 % de los casos de ECJ son de origen genético y hereditario. En 1950, se reportó y se realizó el seguimiento del primer caso familiar con miembros de tres generaciones probablemente afectados.[17] Asimismo, está documentada la transmisión de varón a varón. En 1979, se estableció que cerca de un 15 % de los casos de ECJ son de tipo familiar.[18] En 1981, otro estudio sobre 73 familias determinó un historial consistente con un patrón de herencia autosómica dominante.[19] El fenotipo clínico es semejante al observado en el ECJ esporádico, aunque esta forma suele presentarse a edades más tempranas.[14]
ECJ adquirida
La enfermedad es transmitida por exposición directa al prion, mediante contacto con tejidos cerebrales o del sistema nervioso infectados. Se ha probado el contagio mediante ciertos procedimientos médicos, estando también expuestos los veterinarios que han tenido contacto con vacas u ovinos enfermos, personal de la industria de la carne, etc. Sin embargo, no hay pruebas de que la ECJ pueda contagiarse mediante un contacto casual con los enfermos. Desde que la ECJ se describiera por primera vez, menos de 1 % de los casos se han probado como adquiridos más allá de toda duda.
Si bien la ECJ puede transmitirse de persona a persona, el riesgo de que esto ocurra es sumamente bajo. La ECJ no parece poder transmitirse a través del aire o al tocar a otra persona o mediante la mayoría de las formas de contacto casual. Los cónyuges y otros miembros de la familia de pacientes con ECJ esporádica no están sometidos a un riesgo mayor de contraer la enfermedad que la población en general (excepto en los casos obviamente hereditarios respecto de los hijos y otros descendientes).
- Forma iatrogénica
- El contacto directo o indirecto con el tejido cerebral y el líquido de la médula espinal de los pacientes infectados debe evitarse para impedir la transmisión de la enfermedad a través de estos materiales. En unos cuantos casos muy raros, pero perfectamente demostrados, la ECJ se ha propagado a otras personas a raíz de injertos de duramadre (una de las meninges, tejidos que cubren el cerebro), córneas trasplantadas, implantación de electrodos inadecuadamente esterilizados en el cerebro e inyecciones de hormona somatotropa contaminada obtenida de glándulas pituitarias humanas tomadas de cadáveres.[20][21] Los médicos llaman a estos casos —provocados por procedimientos médicos— "casos iatrogénicos".
- Nueva variante
- La enfermedad de Creutzfeldt-Jakob variante o nueva variante (ECJv o ECJnv) fue descrita en el Reino Unido y en Francia, y comienza principalmente con síntomas psiquiátricos. Afecta a pacientes más jóvenes que los de otros tipos de ECJ y tiene una duración más larga de lo ordinario desde el comienzo de los síntomas hasta la muerte. Fue descubierta en 1996 y es el tipo más relacionado con la exposición al prion responsable del mal de la vaca loca. Se cree que la ECJv es adquirida a partir del ganado infectado con EEB.[22]
Biología molecular
En un principio, se creyó que en el origen de la ECJ y otras EET existía un "virus lento" (Lentivirus) u otro organismo desconocido. Sin embargo, estos nunca han podido ser aislados. Además, el agente que ocasiona la ECJ tiene varias características que son raras en microorganismos tales como los virus y las bacterias. Es inmune a todos los métodos comunes de esterilización, no contiene ninguna información genética en forma de ácidos nucleicos (ADN o ARN) y presenta generalmente un largo periodo de incubación antes de que aparezcan los síntomas. En algunos casos, este lapso puede ser de hasta 40 años. La teoría científica principal —demostrada en la actualidad para el kuru, la EEB y los casos adquiridos de ECJ— afirma que estas EET no son ocasionadas por un microorganismo sino por un tipo de proteína llamado prion e identificado como PrPSc.[4]
Los priones se presentan en forma normal como una proteína inocua hallada en las células del cuerpo,[4] que controla ciertos aspectos de la vida celular. Sin embargo, el prion puede tomar también una forma infecciosa capaz de ocasionar la enfermedad. Este es el motivo de que el sistema inmune no sea capaz de luchar contra el prion, ya que se trata de una proteína propia, cuya presencia es normal en todas las células del cuerpo.
Las formas inocuas e infecciosas de la proteína y el prion son casi idénticas, pero la forma infecciosa adquiere una configuración plegada diferente a la de la proteína normal.[4]
En la ECJ adquirida, el prion ingresa al organismo a través del contacto con priones infecciosos. En la ECJ hereditaria, el gen responsable de producir la proteína normal ha sufrido una mutación tal que sólo es capaz de producir la proteína patológica. Acaso la causa de la inexplicable forma esporádica sea que los priones normales se transforman —por razones aún desconocidas— en la versión infecciosa de la proteína.
La característica más letal de estos priones patológicos es que aunque haya uno solo de ellos, esta única molécula es capaz de "reconfigurar" a sus similares normales, produciendo una especie de imparable reacción en cadena que deja al organismo sin moléculas "sanas". Una vez que aparecen, las proteínas de los priones anormales se unen y forman fibras o acumulaciones llamadas "placas amiloides", que pueden verse al microscopio. Las fibras y las placas pueden comenzar a depositarse años antes de que empiecen a aparecer los síntomas de las ECJ. Todavía no está claro el papel que desempeñan estas estructuras en la enfermedad o cómo pudieran afectar a los síntomas.
En las EET hereditarias, se han identificado varias (hasta 20) mutaciones diferentes en el gen de los priones. La mutación específica que se encuentra en cada familia afecta posiblemente al tipo de EET que experimentará, a la frecuencia con que aparece la enfermedad en la familia y al tipo de síntomas más notables. Sin embargo, no todas las personas con mutaciones en el gen de los priones adquieren la ECJ. Esto indica que las mutaciones pueden meramente aumentar la susceptibilidad a la ECJ y que tal vez existan otros factores aún desconocidos que también desempeñan un papel en la enfermedad, sin descartar el contagio por diversas vías.
Genética
El gen productor del prion ha sido bautizado PRNP (Prion-Related Protein). Este gen se encuentra localizado en el brazo corto (p) del cromosoma 20, en la posición 20pter-p12 (es decir, entre la posición 20p12 y el final o término del brazo), ocupando 15 000 pares de bases: más precisamente, desde el par 4 615 068 al 4 630 233 del cromosoma. La proteína codificada por este gen se denomina PrPC (c por "celular", ya que se halla presente en todas las células). Sin embargo, una mutación en un único punto de este gen puede producir una forma patológica de la misma proteína, que ha sido llamada PrPSc (por scrapie, nombre inglés de la tembladera ovina o caprina —ICTVdb 90.001.0.01.001).
El reemplazo de una molécula del aminoácido lisina por prolina en uno de los sectores del gen PRNP hace que un grupo de ocho aminoácidos (octapéptido) comience a replicarse (copiarse a sí mismo), yendo cada nueva copia a cambiar a otras moléculas de PrPC en PrPSc. De este modo se explica la capacidad del prion para convertir moléculas normales en priones a su vez.
En 1995 se demostró que el error en este único aminoácido altera radicalmente toda la enorme estructura del resto de la proteína, reconfigurándola en una nueva forma denominada prion.
Cinética molecular del prion
Al encontrarse una molécula de PrPsc con una normal, la "moldea" o repliega en una forma diferente a la que tenía, reemplazando el aminoácido necesario para convertirla en una como ella. Esta va a transformar a otras, y así sucesivamente. Este proceso es particularmente fácil y eficiente en las neuronas. Parte de los túneles y vacuolas que dan al cerebro enfermo su característico aspecto de esponja son el resultado del "estallido" de neuronas infectadas que han liberado miles de priones en el medio intercelular.
Las teorías actuales afirman que las células nerviosas poseen un receptor químico en su membrana que se comporta a manera de "cerradura". Ciertas proteínas "asesinas", encargadas de aniquilar a las neuronas enfermas o anormales, introducen parte de su estructura en estos receptores a modo de "llave". El receptor de las células sanas está obstruido por una molécula de PrPc (es decir, normal), de forma que las proteínas asesinas no puedan reconocerla. Si el orificio está libre, la proteína asesina mata a la célula en cuestión.
Sin embargo, el prion, con su estructura deformada, no puede ocupar el sitio que le corresponde en la membrana, como una llave equivocada no puede introducirse en una cerradura ajena. La célula normal, pues, presenta el receptor libre, dando a la proteína asesina la impresión de que se trata de una que debe morir. En la ECJ y las demás EET, las proteínas asesinas matan a las neuronas normales confundiéndolas con células marcadas para ser destruidas.
Enfermedades relacionadas
| EET o enfermedades por priones en animales[14] |
|---|
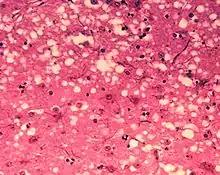 Microfotografía de tejido encefálico en animal con EEB. y túneles que le dan un aspecto de esponja. |
| Enfermedad esporádica, posiblemente genética e infecciosa |
| Scrapie (inglés), tremblante (francés), tembladera, modorra o rascadera (español) Cabras, ovejas, carneros |
| Enfermedad infecciosa, posiblemente esporádica |
| EE bovina o "vacas locas" (vacas y toros) |
| EE felina (gatos en libertad y cautivos) |
| Encefalopatía transmisible del visón, del ciervo y antílopes |
| EE de rumiantes de zoo y ungulados |
| EE en diversos animales de experimentación |
| Enfermedad genética |
| EE de ratón transgénico |
Existen varias otras enfermedades humanas relacionadas con la ECJ, todas ellas producidas por priones similares:
- Kuru: primera enfermedad priónica identificada en el ser humano, al principio se la confundió con una patología virósica. Era común en una tribu aislada de Nueva Guinea, entre cuyos habitantes se acostumbraba comer los cerebros de los familiares muertos. Fue descubierta alrededor de 1900, y, gracias a que los nativos fueron disuadidos de sus extraños hábitos, hoy se la considera extinta.
- Síndrome de Gerstmann-Sträussler-Scheinker (GSS): se trata de un mal rarísimo, que solo afecta a un puñado de familias en todo el mundo. Por este motivo, se considera casi segura la herencia del gen responsable de producir el prion. De síntomas similares a la ECJ, su desarrollo es más lento (cursa entre 2 a 10 años). Es también fatal e incurable.
- Insomnio familiar fatal (IFF): producido por una mutación diferente del gen del prion de la ECJ, es extremadamente raro y sus pocos casos registrados se han observado en España.
- Otras enfermedades priónicas: la variedad de sintomatologías clínicas en las enfermedades priónicas ha llevado a la definición clínica de otras patologías del mismo grupo (incluyendo numerosos casos de Alzheimer que han sido reexaminados y hoy se cuentan entre las EET):
- Demencia por prion sin patología característica
- Demencia con paraparesia espástica
- Demencia talámica
- Encefalopatía espongiforme familiar asociada a una nueva mutación en el gen PrP
- Gliosis subcortical progresiva
- Enfermedad mental sin signos neurológicos
- Enfermedad de Alzheimer familiar producida por priones
Asimismo, se conocen numerosas variantes que afectan a distintas especies animales: équidos, bovinos, ovinos, caprinos, felinos, mustélidos y ungulados silvestres.
Tratamiento
No existe tratamiento que pueda curar la enfermedad. Algunos de sus síntomas, como los espasmos, pueden ser controlados a través de tratamiento paliativo.[23] Los investigadores han sometido a prueba muchos fármacos, entre ellos la amantadina, los esteroides, el interferón, el aciclovir, la clorpromazina,[24] y diversos agentes antivirales y antibióticos. No obstante, ninguno de estos tratamientos ha demostrado ser beneficioso.
El único tratamiento posible de la ECJ tiene como propósito principal aliviar los síntomas hasta donde sea posible y mejorar la calidad de vida del paciente. Las drogas opiáceas pueden ayudar a reducir el dolor si se presenta, y el clonazepam y el valproato de sodio pueden ayudar a paliar el mioclono. Durante las últimas etapas de la enfermedad, cambiar de posición al paciente ayuda a evitar lesiones y escaras, propias de la postración en cama. Puede emplearse un catéter para drenar la orina si el paciente no puede controlar la función de la vejiga y también puede utilizarse alimentación artificial, incluyendo líquidos intravenosos.
Investigación
Muchos investigadores están estudiando la ECJ, tratando de descubrir los factores que influyen en la infectividad de los priones. Utilizando modelos de roedores con la enfermedad y tejido cerebral de autopsias, también están intentando identificar aquellos factores que influyen en la susceptibilidad a la enfermedad y los que gobiernan el curso de la misma cuando aparece. Los estudios para confirmar el diagnóstico son de muy alto costo y solo los países con tecnología de punta pueden realizarlos, así que aun es más difícil hacer el diagnóstico.
Esperan utilizar estos conocimientos para formular mejores pruebas diagnósticas y aprender el mecanismo íntimo por el que el prion mata a las neuronas a fin de que puedan formularse tratamientos eficaces.
El problema de las transfusiones de sangre
Un informe científico publicado en la prestigiosa revista británica The Lancet[25] ha demostrado que la ECJv puede transmitirse a través de las transfusiones de sangre. El descubrimiento alarmó a los sistemas de salud, porque es posible que una gran epidemia de la enfermedad aparezca en el futuro cercano. No existe una prueba que permita determinar si la sangre donada está o no infectada con el prion. Aunque el donante no presente síntomas, puede hallarse en la fase latente de la enfermedad y transmitirla mediante la sangre donada. Como reacción a este informe, el gobierno británico prohibió donar sangre a todos aquellos que hayan recibido una transfusión de sangre en fecha posterior a enero de 1980.
El 28 de mayo de 2002 la Administración de Drogas y Alimentos de los Estados Unidos (FDA), prohibió a su vez donar sangre a las personas que hayan vivido en las zonas de Europa consideradas de alto riesgo de EEB o ECJ entre 1980 y mediados de los 90. Dado el gran número de militares norteamericanos residentes en Europa, se cree que más del 7 % de los mismos se verán impedidos de donar sangre en cumplimiento de esta norma.
Medidas similares fueron adoptadas por la Cruz Roja Australiana, que no permite donar a quienes hayan vivido un tiempo (acumulado) de al menos seis meses en el Reino Unido entre 1980 y 1996. Canadá ha prohibido la donación de sangre a las personas que hayan vivido seis meses o más en el Reino Unido desde 1980. Lo mismo se aplica a quienes hayan residido en Francia por más de seis meses.
Víctimas famosas
La prensa masiva comenzó a ocuparse de la ECJ cuando falleció víctima de la misma el célebre coreógrafo ruso-estadounidense George Balanchine, a quien se considera la primera víctima famosa de este mal. El artista nacido en San Petersburgo comenzó experimentando extraños problemas de equilibrio mientras danzaba en 1978. Después de perder la vista y el oído, quedó completamente incapacitado hacia 1982. Posteriormente desarrolló angina de pecho y un grave ataque cardíaco que obligó a practicarle una cirugía de baipás coronario. Ciego, sordo y paralítico, falleció el 30 de abril de 1983. La autopsia de su cerebro demostró claramente que había muerto de Creutzfeldt-Jakob.
El oncólogo Josep Baselga también murió debido a esta patología
Véase también
Notas
- Cabe señalar que en español a veces se utiliza este término de manera libremente intercambiable con la expresión "Síndrome de Creutzfeldt-Jakob", lo que puede ser motivo de confusión.
Referencias
- La enfermedad de Creutzfeldt-Jakob. The Institutes. 2000. Consultado el 12 de abril de 2022.
- Henry, Ronnie; Murphy, Frederick A. (2017-06). «Etymologia: Creutzfeldt-Jakob Disease». Emerging Infectious Diseases 23 (6): 956-956. ISSN 1080-6040. PMC 5443459. doi:10.3201/eid2306.ET2306. Consultado el 29 de mayo de 2022.
- Ritchie, Diane L.; Ironside, James W. (1 de enero de 2017). Legname, Giuseppe, ed. Neuropathology of Human Prion Diseases. Prion Protein (en inglés) 150. Academic Press. pp. 319-339. doi:10.1016/bs.pmbts.2017.06.011. Consultado el 29 de mayo de 2022.
- Tratado de Neurología Clinica. Editorial Universitaria. 2005. ISBN 978-956-11-1798-3. Consultado el 12 de abril de 2022.
- «The Nobel Prize in Physiology or Medicine 1997». NobelPrize.org (en inglés estadounidense). Consultado el 29 de mayo de 2022.
- El Control de las enfermedades transmisibles, 18a. Edición. Pan American Health Org. 2005. p. 151. ISBN 978-92-75-31613-9. Consultado el 12 de abril de 2022.
- Brown P, Cathala F, Castaigne P, Gajdusek DC. "Creutzfeldt-Jakob disease: clinical analysis of a consecutive series of 230 neuropathologically verified cases". Ann Neurol. 1986 nov;20(5):597-602. PMID 3539001
- Uttley, Lesley; Carroll, Christopher; Wong, Ruth; Hilton, David A; Stevenson, Matt (2020-01). «Creutzfeldt-Jakob disease: a systematic review of global incidence, prevalence, infectivity, and incubation». The Lancet Infectious Diseases 20 (1): e2-e10. ISSN 1473-3099. doi:10.1016/s1473-3099(19)30615-2. Consultado el 29 de mayo de 2022.
- Sitammagari, Kranthi K.; Masood, Wajeed (2022). Creutzfeldt Jakob Disease. StatPearls Publishing. PMID 29939637. Consultado el 29 de mayo de 2022.
- Belay, Ermias D.; Schonberger, Lawrence B. (2002-12). «Variant Creutzfeldt-Jakob disease and bovine spongiform encephalopathy». Clinics in Laboratory Medicine (en inglés) 22 (4): 849-862. doi:10.1016/S0272-2712(02)00024-0. Consultado el 3 de junio de 2022.
- Uttley, Lesley; Carroll, Christopher; Wong, Ruth; Hilton, David A; Stevenson, Matt (2020-01). «Creutzfeldt-Jakob disease: a systematic review of global incidence, prevalence, infectivity, and incubation». The Lancet Infectious Diseases 20 (1): e2-e10. ISSN 1473-3099. doi:10.1016/s1473-3099(19)30615-2. Consultado el 1 de junio de 2022.
- Ruiz, Vicente Ausina; Guillén, Santiago Moreno (20 de julio de 2006). Tratado SEIMC de enfermedades infecciosas y microbiología clínica. Ed. Médica Panamericana. p. 1000. ISBN 978-84-7903-921-9. Consultado el 12 de abril de 2022.
- Will RG, et al. "A new variant of Creutzfeldt-Jakob disease in the UK". Lancet. 1996 abr 6;347(9006):921-5. PMID 8598754
- Bermejo FP, Muñoz D. "Encefalopatías espongiformes transmisibles (EET) o enfermedades producidas por priones". Rev Adm Sanitaria 2001;17:27-44.
- Variant Creutzfeldt-Jakob disease, current data. 21 de julio de 2012. Archivado desde el original el 21 de julio de 2012. Consultado el 29 de mayo de 2022.
- Tyler KL. "Creutzfeldt-Jakob disease". N Engl J Med. 2003 feb 20;348(8):711-9. PMID 12594311
- Jakob H, Pyrkosch W, Strube H. "Hereditary form of Creutzfeldt-Jakob disease (Backer family)". Arch. Psychiat. 1950;184(7):653-74. PMID 15433375
- Masters CL, Harris JO, Gajdusek DC, Gibbs CJ Jr, Bernoulli C, Asher DM. "Creutzfeldt-Jakob disease: patterns of worldwide occurrence and the significance of familiar and sporadic clustering". Ann Neurol. 1979 feb;5(2):177-88. PMID 371520
- Masters CL, Gajdusek DC, Gibbs CJ Jr. "The familial occurrence of Creutzfeldt-Jakob disease and Alzheimer's disease". Brain. 1981 sep;104(3):535-58. PMID 7023604
- Brown P, Preece MA, Will RG. "Friendly fire in medicine: hormones, homografts, and Creutzfeldt-Jakob disease". Lancet. 1992 jul 4;340(8810):24-7. PMID 1351607
- Brown P, Cervenakova L, Goldfarb LG, McCombie WR, Rubenstein R, Will RG, Pocchiari M, Martínez-Lage JF, Scalici C, Masullo C, et al. "Iatrogenic Creutzfeldt-Jakob disease: an example of the interplay between ancient genes and modern medicine." Neurology. 1994 feb;44(2):291-3. PMID 8309577
- Collinge J, Sidle KCL, Heads J, Ironside J, Hill AF. "Molecular analysis of prion strain variation and the aetiology of 'new variant' CJD." Nature. 1996 oct 24;383(6602):685-90. PMID 8878476
- Manix, Marc; Kalakoti, Piyush; Henry, Miriam; Thakur, Jai; Menger, Richard; Guthikonda, Bharat; Nanda, Anil (1 de noviembre de 2015). «Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy». Neurosurgical Focus (en inglés estadounidense) 39 (5): E2. ISSN 1092-0684. doi:10.3171/2015.8.FOCUS15328. Consultado el 10 de julio de 2023.
- Korth C, et al. "Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease." Proc Natl Acad Sci U S A. 2001 ag 14;98(17):9836-41. PMID 11504948
- Peden AH, et al. "Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient." Lancet. 2004 ag 7-13;364(9433):527-9. PMID 15302196
Bibliografía
- López-Herrera A, et al. "El desafío de las enfermedades priónicas". Vet Méx. 2002;33(4). Disponible en línea (PDF) Archivado el 26 de junio de 2020 en Wayback Machine.
- McKintosh E, Tabrizi SJ, Collinge J. "Prion diseases". Journal of NeuroVirology. 2003;9:183–93. Disponible en línea (PDF)
Enlaces externos
- Cronología de la nueva variante de la enfermedad de Creutzfeldt-Jakob Por Joaquín Escudero Torrella. Sección de Neurología. Hospital General de Castellón.
- Demencia vascular y otras enfermedades que cursan con demencia Por F. Bermejo Pareja y otros. Medicine. 14 abr 2003;8(101):5453-64.
- Clínica de las enfermedades priónicas Servicio de Neurología del Hospital General de Elche.
- Sistema de Información sobre Enfermedades Raras
- Alzheimer Europe Información sobre el Alzheimer, la ECJ y otros tipos de demencia.
- "Destructores de cerebros", artículo informativo y didáctico sobre las enfermedades producidas por priones
- En inglés
- NCJDSU Unidad Nacional de Vigilancia de la ECJ del Reino Unido.
- Genetics Home Reference Enfermedades priónicas: variedad de recursos informativos; contenidos producidos por los National Institutes of Health (publicados bajo dominio público).
- Creutzfeldt-Jakob Disease Information Page NINDS
- MedlinePlus Enciclopedia 000788
| Control de autoridades |
|
|---|
 Datos: Q49989
Datos: Q49989 Multimedia: Creutzfeldt–Jakob disease / Q49989
Multimedia: Creutzfeldt–Jakob disease / Q49989