Esclerosis sistémica
La esclerosis sistémica o esclerodermia sistémica (gr. "piel endurecida") es un término que abarca un espectro de enfermedades autoinmunes del tejido conjuntivo que involucran cambios en la piel, los vasos sanguíneos, los músculos y los órganos internos.[1]
| Esclerosis sistémica | ||
|---|---|---|
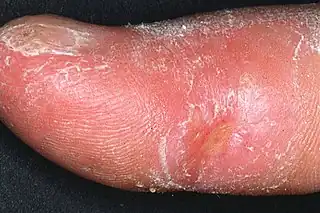 Pulgar de un paciente con esclerosis sistémica | ||
| Especialidad | reumatología | |
| Sinónimos | ||
| Esclerodermia sistémica | ||
Se desconocen las causas de esta enfermedad. Podría tratarse de un conjunto de enfermedades que afectan el tejido conjuntivo del cuerpo, que hacen que se endurezca y se ponga grueso. Este tejido le da soporte a la piel y a los órganos internos.
Es un tipo de trastorno autoinmunitario, una afección que ocurre cuando el sistema inmunitario ataca por error y destruye tejido corporal sano.[1]
Clasificación
La esclerosis sistémica o esclerodermia sistémica es una enfermedad autoinmune multi-sistémica, en la que hay un aumento de actividad de los fibroblastos, resultando en un crecimiento anormal de tejido conectivo. Esto provoca daño vascular y fibrosis (tejido cicatricial). La fibrosis ocurre en la piel, el tracto gastrointestinal y otros órganos internos, como el corazón, los pulmones o los riñones. Cuando el tejido normal de un órgano se reemplaza por tejido fibroso, dicho órgano deja de funcionar adecuadamente.[2]
Una vez diagnosticada, lo primordial es detectar precozmente la presencia de afectación visceral.
Se clasifica a su vez en dos tipos:[3]
Esclerosis/esclerodermia sistémica con afectación cutánea limitada
- 70% de los casos de esclerosis sistémica.
- Presenta afectación dérmica (de la piel) limitada. Afecta solo la cara, antebrazos y piernas hasta la rodilla.
- El término más antiguo para la esclerosis sistémica con afectación cutánea limitada es el síndrome de CREST. CREST es un acrónimo de los nombres en inglés de los cinco rasgos característicos clínicos principales:
- Calcinosis (‘calcificación’ (depósitos de calcio)).
- Raynaud's syndrome (‘síndrome de Raynaud’).
- Esophageal dysmotility (‘dismotilidad esofágica’ (disfunción esofágica)).
- Sclerodactyly (‘esclerodactilia’).
- Telangiectasia (‘telangiectasia’ (dilatación de pequeños vasos)).
Esclerosis/esclerodermia sistémica con afectación cutánea difusa
- 30% de los casos de esclerosis sistémica.
- Con afectación dérmica difusa. Involucra también los brazos, los muslos o el tronco.
Esclerosis sistémica sin esclerodermia
Abarca los subtipos de la enfermedad actualmente aceptados.[4]
Existen algunos subtipos raros en los que se produce afectación de órganos internos sin cambios en la piel.
Epidemiología
Se presenta a lo largo del mundo, en todos los grupos étnicos. Las cifras de incidencia y prevalencia varían ampliamente y parece que hay una gran variación geográfica.
Afecta generalmente a personas de entre 30 y 50 años, pero puede presentarse en cualquier grupo de edad. Es rara en los niños.
Las mujeres a menudo se ven más afectadas que los hombres, en una proporción de 4-5:1.
Etiología
Las características principales son la sobreproducción y el depósito excesivo de colágeno, el daño vascular y la inflamación o la autoinmunidad.[2] Se desconocen las causas de este padecimiento, pero existen diversas teorías.
La teoría autoinmune sugiere que el propio sistema inmunitario es parcialmente responsable. Habitualmente, el sistema inmune del cuerpo produce unas sustancias químicas en la sangre llamadas citoquinas que, actuando como señales, coordinan la defensa del cuerpo contra las bacterias, contra los virus y contra otros invasores extraños. Hay numerosas teorías que hablan de una activación inapropiada del sistema inmunitario, causando niveles anormales de citoquinas. Estas, a su vez, preparan un ataque no contra invasores extraños, sino contra los tejidos sanos del propio cuerpo, estimulando la sobreproducción de colágeno.
La teoría vascular implica a los vasos sanguíneos. Los vasos sanguíneos dañados, especialmente los pequeños, son típicos en la esclerodermia. Este daño provoca su estrechamiento y endurecimiento, y los induce a reaccionar al frío o al estrés. Estas reacciones pueden causar más daño a los propios vasos y a los órganos a los que suministran.
También podría haber una conexión entre la construcción del exceso de colágeno y el cambio de los vasos sanguíneos. Ya hay en marcha estudios para determinar exactamente cuáles son los factores causantes de los daños a los vasos sanguíneos, los procesos que tienen lugar y su significado, para su prevención y tratamiento. Se están realizando investigaciones para estudiar estas y otras teorías. Se espera que una mejor comprensión de las causas lleve a mejores métodos de tratamientos, y finalmente, a la cura.[5]
Es probable que haya una predisposición genética. Los factores ambientales pueden jugar un papel en el desencadenamiento de la enfermedad. Estos pueden incluir los virus (como el citomegalovirus), productos químicos (tales como cloruro de vinilo, algunos pesticidas, derivados del benceno y sílice), las drogas (como cocaína, supresores del apetito, la penicilamina y la vitamina K).[2]
Cuadro clínico
Los síntomas de presentación más comunes son el fenómeno de Raynaud (que puede preceder a otros síntomas por algunos años), el endurecimiento de piel de las manos o la cara y síntomas esofágicos.[3]·[6]
Los primeros síntomas pueden ser inespecíficos (por ejemplo, fatiga, dolores osteomusculares e hinchazón de las manos). Tanto la esclerosis sistémica con afectación cutánea limitada como la difusa pueden afectar los órganos internos y la gravedad de los cambios de la piel no necesariamente refleja la gravedad de la afectación de órganos internos.[3]·[6]
Esclerosis/esclerodermia sistémica con afectación cutánea limitada
- Por lo general una enfermedad leve, con menos participación de la piel, aparición y progresión lentas.
- El inicio lento puede hacer que los síntomas pasen relativamente desapercibidos hasta que ocurran complicaciones internas.
Esclerosis/esclerodermia sistémica con afectación cutánea difusa
- Por lo general, con un inicio más rápido, con engrosamiento de la piel y fenómeno de Raynaud presentándose asociados o dentro de un corto intervalo de tiempo. Los cambios en la piel pueden propagarse rápidamente, a los pocos meses del inicio de la enfermedad.
- Los cambios en la piel pueden remitir después de varios años, con ablandamiento de la piel y mejora significativa de la movilidad.
- Los síntomas tienden a ser peores en los primeros 3 a 5 años de la enfermedad, después de lo cual hay una fase estable y un mayor deterioro es poco probable. La enfermedad entonces puede invertir en cierta medida, con el reblandecimiento de la piel y mejora de la movilidad.
- La afectación de órganos internos es más común.
Características generales
- Fatiga.
- Pérdida de peso.
Características de la piel
- Signos en las manos:
- - Hinchazón de los dedos de las manos y de los pies. Es un signo temprano común. Los dedos tienen aspecto de salchicha y el movimiento de las manos puede ser limitado.
- - La piel se vuelve dura y engrosada. Esto puede limitar el movimiento articular o causar contracturas de las articulaciones. En los dedos, se conoce como esclerodactilia.
- - La hinchazón y esclerosis puede reducir el movimiento de las manos, de manera que los pacientes pueden ser incapaces de cerrar el puño o para colocar las superficies de las palmas juntas en “signo de oración”.
- - Las yemas de los dedos pueden presentar úlceras, cicatrices puntiformes o pérdida de masa en los pulpejos.
- - Fenómeno de Raynaud. Este es el síntoma más común y está presente en algún momento en el 90 % de los casos. El fenómeno de Raynaud con dedos hinchados se consiera que es el signo principal que indica una probable esclerosis sistémica.[7]
- Calcinosis. Nódulos o bultos de material calcáreo, que pueden atravesar la piel.
- Cara y boca:
- - Tirantez de la piel del rostro.
- - Labios apretados (microstomía). Puede dificultar la higiene dental.
- Apariencia de la piel “sal y pimienta”, debido a las zonas de hipo e hiperpigmentación.
- Sequedad o picazón en la piel. Reducción del vello o cabello sobre las áreas afectadas de la piel.
Características del aparato locomotor
- Inflamación y dolor articular.
- Restricción de los movimientos articulares, contracturas y atrofia muscular debidos a la esclerosis de la piel.
- Fricción de los tendones. Palpables/audibles sobre los tendones de los flexores y extensores de las manos, rodillas y tobillos.
Características gastrointestinales
- Acidez gástrica y esofagitis por reflujo.
- Cicatrices y disfagia esofágica.
- Vaciamiento gástrico retrasado. Por ejemplo, sensación de plenitud después de las comidas.
- Reducción de la motilidad del intestino delgado. Puede causar sobrecrecimiento bacteriano, con hinchazón, malabsorción, diarrea y desnutrición.
- Estreñimiento debido a la reducción de la motilidad colónica.
Funciones pulmonares
- La fibrosis pulmonar (enfermedad pulmonar intersticial):
- - Se produce en hasta el 75 % de los pacientes con esclerodermia, pero solo unos pocos desarrollan la enfermedad en etapa terminal.[8]
- - Causa enfermedad pulmonar restrictiva.
- - Síntomas y signos: disnea de esfuerzo, tos, crepitantes.
- - Ocurre en aproximadamente el 10-15 % de los pacientes con esclerodermia.[3]
- - Es una de las principales causas de muerte en la esclerosis sistémica. La presencia de hipertensión arterial pulmonar reduce drásticamente la tasa de supervivencia.
- - Síntomas y signos: disnea de esfuerzo, síncope, características de deformación del ventrículo derecho.
- - Investigaciones recientes han tratado de definir las herramientas de detección. Estas incluyen la supervisión de la función del pulmón, electrocardiograma (ECG), niveles de urato en sangre, y prohormona N-terminal del péptido natriurético cerebral (NT-proBNP); y teniendo en cuenta la presencia del anticuerpo anti-centrómero y la historia o presencia de telangiectasia.[9]
Diagnóstico
El diagnóstico diferencial de la incluye el escleredema clásico de Buschke, el escleromixedema, la enfermedad injerto contra huésped y la porfiria cutánea tarda, todas con contexto clínico específico que las diferencia fácilmente.
El diagnóstico puede difícil en las primeras etapas pero inequívoco la enfermedad está avanzada. La presencia de Raynaud acompañado de capilaroscopia patológica y/o distintos anticuerpos en la sangre, específicos de ES llamados anticuerpos antinucleares (ANA), pueden dar el diagnóstico de una enfermedad en sus comienzos.
El examen periódico del grado y extensión de la lesión cutánea (total skin score) es el mejor método para detectar pacientes con riesgo de desarrollar afectación visceral.
El examen manual de la piel es más sensible que la biopsia cutánea y supone el diagnóstico definitivo en más del 90% de los casos.
El diagnóstico lo suelen dar los médicos con amplia experiencia en el tratamiento de esta enfermedad teniendo en cuenta lo siguiente: el historial médico, incluyendo los síntomas pasados y los del presente; un minucioso examen físico y pruebas realizadas en una gran variedad de tests y otros estudios. Al hacer el diagnóstico, es importante no solo confirmar la presencia de la esclerodermia, sino también su alcance y gravedad, pues hay que considerar la implicación de los órganos internos.[5]
Criterios diagnóstico
Los criterios diagnósticos y de clasificación se encuentran en proceso de revisión, en aras de incluir y recoger las fases tempranas. Desde 1980, los criterios de diagnóstico han sido esclerodermia proximal (próxima a la articulación metacarpofalángica), esclerodactilia, cicatrices puntiformes de pulpejos o pérdida de la pulpa y fibrosis pulmonar bibasal.
Sin embargo, en 2013, con la colaboración del Colegio Americano de Reumatología y la Liga Europea Contra el Reumatismo (ACR / EULAR) fue propuesto un nuevo conjunto de criterios.[10]
A otros elementos se les da una puntuación ponderada y una puntuación de 9 o más significa el diagnóstico de esclerosis sistémica. Al tradicional criterio mayor del engrosamiento de la piel que se extiende próxima a las articulaciones metacarpofalángicas se le da una puntuación de 9 y, por lo tanto, es suficiente por sí solo para hacer un diagnóstico.
Las siguientes características se incluyen en el nuevo sistema:
- Engrosamiento de la piel que se extiende proximal a las articulaciones metacarpofalángicas (Puntuación 9).
- Engrosamiento de la piel de los dedos (Puntuación 2 para los dedos hinchados, 4 para esclerodactilia).
- Lesiones en la yema del dedo (Puntuación 2 para las úlceras, 3 para las cicatrices puntiformes de pulpejos).
- Telangiectasia (Puntuación 2).
- Hipertensión arterial pulmonar y/o enfermedad pulmonar intersticial (Puntuación 2).
- Fenómeno de Raynaud (Puntuación 3).
- Autoanticuerpos relacionados con esclerosis sistémica (puntuación 3).
Diagnóstico precoz: la iniciativa VEDOSS
La iniciativa VEDOSS (= Very Early Diagnosis Of Systemic Sclerosis) en Europa[11] identifica las siguientes características como la clave para el diagnóstico de la esclerosis sistémica en una etapa muy temprana:
- Anticuerpos antinucleares (ANAs).
- Anticuerpos específicos de la esclerodermia.
- Patrón de esclerosis sistémica en la capilaroscopia del lecho ungueal.
- Dedos hinchados en los pacientes con Síndrome de Raynaud.
Análisis de sangre
Los parámetros a evaluar en sangre incluyen:[6]
- Hemograma completo.
- Velocidad de sedimentación globular (VSG) y proteína C reactiva (PCR).
- Bioquímica completa y función renal.
- Cuantificación de inmunoglobulina IgA.
- Anticuerpos antinucleares (ANA).
Otras pruebas
- Proteinograma urinario. Como base de referencia de si hay complicaciones renales.
- Biopsia de piel. Puede ayudar al diagnóstico.
- Capilaroscopia periungueal. Ayuda a evaluar la probabilidad de esclerodermia en pacientes con fenómeno de Raynaud o dedos hinchados. También es útil en la predicción del riesgo de desarrollar úlceras.[12]
- Radiografía de la mano. Puede mostrar calcinosis.
- Termografía con provocación con frío. Ayuda a evaluar la gravedad del fenómeno de Raynaud.
Seguimiento
El seguimiento y las revisiones regulares están dirigidos a la detección temprana y al tratamiento de las complicaciones. Incluyen:[6]
- Revisión periódica de los síntomas.
- Control de la presión arterial.
- Monitorización de la función renal.
- Pruebas de función pulmonar y tomografía computarizada del tórax.
- Ecocardiograma (ECG) y ecocardiografía.
Tratamiento
No hay cura para la esclerosis sistémica y el tratamiento consiste en controlar los síntomas y prevenir complicaciones.[3]·[6]
Tratamientos no farmacológicos
- Participación y educación del paciente. Conciencia de los problemas urgentes como la crisis renal o síntomas de obstrucción intestinal.
- Fisioterapia para promover la movilidad de las articulaciones y la fuerza muscular.
- Ejercicios en el domicilio para mantener la amplitud de movimientos de la boca, cara, manos...
- Evitar el tabaco y mantener un peso saludable.
- Asesoramiento nutricional y suplementos en caso necesario.
- Para el fenómeno de Raynaud:
- - Prevención: evitar el frío y los traumatismos; usar ropa de abrigo o ropa caliente.
- - En el caso de un ataque: calentar el cuerpo, las manos y los pies suavemente (la piel puede estar adormecida y puede no sentir si la fuente de calor es demasiado caliente); utilizar movimientos suaves del brazo o un masaje suave para ayudar a restablecer la circulación.
- Terapeutas ocupacionales para las adaptaciones en la vida diaria.
- Ayuda cosmética en los casos con cambios en el aspecto de la piel.
Inmunoterapia
La inmunoterapia incluye las siguientes posibilidades:[13]
- Inmunosupresores no selectivos. La ciclofosfamida es la más comúnmente utilizada y se ha asociado con mejoras tanto en la función pulmonar como en la afectación de la piel, pero su eficacia se puede reducir a partir de los dos años de tratamiento. El micofenolato de mofetilo también se ha utilizado, con mejoras en la función pulmonar. La azatioprina y el metotrexato solos pueden no ser tan beneficiosos, pero han demostrado ser eficaces para la afectación de la piel.
- La inmunoterapia de células T específica. Las células T juegan un papel central en gran parte de la patología de la esclerodermia, lo que probablemente explica por qué la ciclosporina es eficaz. Otra inmunoterapia de células T que está siendo probada incluye sirolimus, la globulina antitimocítica y basiliximab.
- Corticosteroides orales.
- Otros tratamientos médicos. El rituximab, las inmunoglobulinas intravenosas y el factor de necrosis tumoral alfa (anti-TNFα) también están siendo investigados para el tratamiento de la esclerodermia.
Tratamiento de los problemas cutáneos
Síntomas del fenómeno de Raynaud y úlceras:[14]
- La nifedipina.
- Otros tratamientos para el fenómeno de Raynaud, que pueden ser eficaces pero aún no están autorizados en algunos países:
- - Nitroglicerina en ungüento.
- - Inhibidores de la fosfodiesterasa 5, ya que son vasodilatadores bien tolerados.
- - Las prostaglandinas. El iloprost por vía intravenosa se recomienda para los síntomas severos en Europa.[15]
- - El losartán, que puede ser más eficaz que la nifedipina.
- Para las úlceras isquémicas:
- - Apósitos simples de prevención.
- - Antibióticos, si están infectadas.
- - Los vasodilatadores pueden ayudar en algunos casos. Por ejemplo, el bosentán puede reducir la aparición de nuevas úlceras, y está recomendado por la EULAR cuando la nifedipina y las prostaglandinas son ineficaces.
Sequedad de la piel o picazón:
- Emolientes.
- Esteroides tópicos (curso corto) o antihistamínicos.
Engrosamiento de la piel:
- Para los pacientes con esclerosis sistémica con afectación cutánea difusa de progresión rápida, considerar ensayar un tratamiento inmunosupresor. Por ejemplo, micofenolato o ciclofosfamida.
Tratamiento de los síntomas musculoesqueléticos
Procedimientos quirúrgicos para indicaciones específicas, tales como:
- Liberación de las contracturas.
- Eliminación de calcinosis problemática.
Mialgia, artralgia y edema doloroso:
- Medicamentos antinflamatorios no esteroideos (AINES), si son tolerados.
- Analgésicos simples.
Tratamiento de los síntomas gastrointestinales
Síntomas del tracto gastrointestinal superior:[3]
- Mantener una postura erguida después de las comidas; elevar la cabecera de la cama; limitar el consumo de alcohol.
- Inhibidores de la bomba de protones.
- También pueden ser necesarios antagonistas de los receptores H2 y agentes pro-motilidad (metoclopramida o domperidona).
- Dilatación de estenosis esofágicas, en caso necesario.
Para el sobrecrecimiento bacteriano intestinal y malabsorción:[3]
- Antibióticos cíclicos.
- Asesoramiento nutricional y suplementos nutricionales. Rara vez, se necesita nutrición parenteral.
Para el estreñimiento:[3]
- Fibra dietética y una buena ingesta de líquidos.
- Laxantes de ablandamiento (tales como lactulosa) y /o fibra soluble (como isphagula o plantago ovata).
Tratamiento de la enfermedad pulmonar
Fibrosis pulmonar (enfermedad pulmonar intersticial):[3]
- Los beneficios de la ciclofosfamida parecen claros, pero se deben sopesar los efectos secundarios.[15]
- Tratamiento de apoyo: tratamiento oportuno de las infecciones de pecho; oxígeno en caso necesario.
Hipertensión arterial pulmonar (HTP):[3]
- El tratamiento farmacológico de la HTP ha mejorado recientemente e incluye:
- - Antagonistas de los receptores de la endotelina. Por ejemplo, bosentán o sitaxentan.
- - Vasodilatadores. Por ejemplo, sildenafil.
- - Derivados de la prostaglandina. Por ejemplo, iloprost (nebulizados o por vía intravenosa) o epoprostenol (infusión)[15]
- Tratamiento de apoyo. Por ejemplo, oxígeno.
Manejo social
El diagnóstico de la Esclerodermia sistémia, puesto que es una enfermedad crónica, produce una distorsión en la vida de los pacientes que repercute a nivel personal, familiar y social. A nivel social los problemas que surgen son entre otros:
- Cambio o pérdida del estatus social por la pérdida o cambio de trabajo. Se pasa a la situación de pensionista por invalidez total y en muchas ocasiones es muy difícil obtener una invalidez absoluta por la falta de información sobre el alcance de la enfermedad.[16]
- Cambio en la utilización del tiempo libre.[16]
- Incremento del gasto por las necesidades propias de la enfermedad.[16]
Complicaciones
Pueden aparecer diversas complicaciones.[3]·[6]
Complicaciones gastrointestinales
- Desnutrición debido a problemas de deglución y otros problemas digestivos.
- Ectasia vascular antral gástrica (“watermelon stomach” o “estómago en sandía”):
- - Puede causar anemia y sangrado gastrointestinal.
- - Es posible que necesite coagulación endoscópica con láser para prevenir el sangrado.
- Obstrucción y pseudo-obstrucción:
- - Puede ocurrir debido a la reducción de la motilidad y al sobrecrecimiento bacteriano.
- - Puede haber complicación por perforación y peritonitis.
- - La pseudo-obstrucción se trata inicialmente con reposo intestinal y antibióticos.
- - Puede ser necesaria una laparotomía.
- Disfunción ano-rectal:
Crisis de esclerodermia renal
- Es una complicación grave con características hipertensivas.
- Puede conducir a la insuficiencia renal si no se trata rápidamente.
- Se presenta en el 5-10% de los pacientes con esclerosis sistémica y es más común en las personas con enfermedad difusa o rápidamente progresiva.[17]
- Presentación:
- - Por lo general, se presenta con hipertensión y oliguria, dolor de cabeza, fatiga, edema, rápido aumento de los niveles de creatinina sérica, proteinuria y hematuria microscópica.
- - Crisis de esclerodermia renal pueden ocurrir con presión arterial aparentemente normal, pero la presión arterial es superior a los valores de referencia (de ahí la importancia de un seguimiento regular de la presión arterial).
- El tratamiento es con enzima convertidora de angiotensina (ECA), además de diálisis en caso necesario.[15]
Complicaciones pulmonares
- Neumonía por aspiración de reflujo severo.
- Debilidad muscular respiratoria si hay miositis severa o enfermedad de la piel extensa que involucra al pecho.
- Neumotórax.
Complicaciones cardíacas
Diferentes anormalidades cardiacas pueden estar asociados con la esclerosis sistémica, que incluyen:[18]
- Enfermedad microvascular de la arteria coronaria (con isquemia miocárdica resultante).
- Fibrosis miocárdica.
- Disfunción sistólica ventricular izquierda y disfunción diastólica ventricular izquierda.
- Pericarditis o derrame pericárdico, que pueden causar insuficiencia cardíaca o insuficiencia cardíaca congestiva.
- Arritmias y trastornos de la circulación (incluyendo bradiarritmias y taquiarritmias).
La amplia variedad de anormalidades hace que sea difícil evaluar la prevalencia. Es probable que las tasas de afectación cardíaca subclínica sean muy elevadas.
El tratamiento es de acuerdo con las características clínicas.
Otras complicaciones
- Disfunción eréctil.[19]
- Depresión.
- Osteoporosis debida a la reducción del flujo sanguíneo.
- Se puede asociar hipotiroidismo.
Síndrome de Sjögren
El Síndrome de Sjögren puede ocurrir en pacientes con un "síndrome de superposición", donde hay características tanto esclerodermia como de Síndrome de Sjögren.
Los síntomas más comunes son sequedad de los ojos y la boca; otras membranas mucosas (por ejemplo, la vagina) pueden ser sintomáticas.
Puede causar irritación de los ojos, disfagia, disfonía y el aumento de caries dentales.
Se trata con Fibrosis miocárdica (por ejemplo, lágrimas artificiales y saliva) y cuidado dental.
Pronóstico
El curso de la enfermedad varía con cada individuo. El pronóstico depende de la magnitud de las complicaciones. Por lo tanto, las cifras de mortalidad varían enormemente. En términos generales, la supervivencia a 10 años es del 60 a 70%.[20] Las muertes por la enfermedad renal se han reducido en los últimos años y la mayor mortalidad es causada por complicaciones graves cardíacas o pulmonares.
Los individuos con esclerosis sistémica con afectación cutánea limitada tienen un pronóstico relativamente positivo. Usualmente morirán por causas naturales o de otra enfermedad, pero no de esclerodermia.
Aquellos en los que el compromiso de piel y órganos es muy importante (sistémico) tienen un pronóstico negativo. Más mujeres que hombres padecen esclerodermia, pero la enfermedad tiene un mayor impacto en los hombres. Luego del diagnóstico, dos tercios de los pacientes viven por lo menos 11 años. Cuanto mayor sea la edad del paciente en el momento del diagnóstico, más probable es que mueran por la enfermedad.
Los pacientes en los que la enfermedad sea severa y progrese rápidamente (un grupo que conforma menos del 10% del total de los pacientes con esclerosis sistémica con afectación cutánea difusa) tienen un 50% de probabilidades de sobrevivir cinco años.[21]
Véase también
Bibliografía
Referencias
- Ariel D. Teitel, MD, MBA, Clinical Associate Professor of Medicine, NYU Langone Medical Center. Review provided by VeriMed Healthcare Network. Also reviewed by A.D.A.M. Health Solutions, Ebix, Inc., Editorial Team: David Zieve, MD, MHA, Bethanne Black, Stephanie Slon, and Nissi Wang. Traducción y localización realizada por: DrTango, Inc. «Esclerodermia» (en spa).
- Ngan, Vanessa. «Systemic sclerosis». DermNet NZ (en inglés).
- Scleroderma Society (Revised October 2008). «Understanding and managing scleroderma». Archivado desde el original el 2 de junio de 2014.
- Fett, N (2013 Jul-Aug). «Scleroderma: nomenclature, etiology, pathogenesis, prognosis, and treatments: facts and controversies». Clin Dermatol 31 (4): 432-7.
- Página oficial Archivado el 7 de julio de 2017 en Wayback Machine. de la enfermedad en la Argentina
- Hinchcliff, M; Varga, J (2008 Oct15). «Systemic sclerosis/scleroderma: a treatable multisystem disease». Am Fam Physician 78 (8): 961-8.
- Minier, T; Guiducci, S; Bellando-Randone, S; et al. (2013 Aug 12). «Preliminary analysis of the Very Early Diagnosis of Systemic Sclerosis (VEDOSS) EUSTAR multicentre study: evidence for puffy fingers as a pivotal sign for suspicion of systemic sclerosis». Ann Rheum Dis.
- Bussone, G; Mouthon, L (2011 Mar). «Interstitial lung disease in systemic sclerosis». Autoimmun Rev 10 (5): 248-55.
- Coghlan, JG; Denton, CP; Grunig, E; et al. (18 de mayo de 2013). «Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study». Ann Rheum Dis.
- van den Hoogen, F; Khanna, D; Fransen, J; et al. (2013 Nov1). «2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative». Ann Rheum Dis 72 (11).
- Muller-Ladner, U; Tyndall, A; Czirjak, L; et al. (2013 Oct 11). «Ten years EULAR Scleroderma Research and Trials (EUSTAR): what has been achieved?». Ann Rheum Dis.
- Jung, P; Trautinger, F (2013 Aug). «Capillaroscopy». J Dtsch Dermatol Ges 11 (8): 731-6.
- Manno, R; Boin, F (2010 Nov). «Immunotherapy of systemic sclerosis». Immunotherapy 2 (6): 863-78.
- Goundry, B; Bell, L; Langtree, M; et al. (2012 Feb). «Diagnosis and management of Raynaud's phenomenon». BMJ 7: 344:e289.
- Kowal-Bielecka, O; Landewe, R; Avouac, J; et al. (2009 May). «EULAR recommendations for the treatment of systemic sclerosis: a report from the EULAR Scleroderma Trials and Research group (EUSTAR)». Ann Rheum Dis 68 (5): 620-8.
- Dr. Adolfo de la Peña Llerandi (2008). «Esclerodermia».
- Denton, CP; Lapadula, G; Mouthon, L; et al. (2009 Jun). Renal complications and scleroderma renal crisis. Rheumatology (Oxford) 48 (Suppl 3).
- Desai, CS; Lee, DC; Shah, SJ (2011 Nov). «Systemic sclerosis and the heart: current diagnosis and management». Curr Opin Rheumatol 23 (6): 545-54.
- Seftel, AD (2012 Aug). «Re: Erectile dysfunction is frequent in systemic sclerosis and associated with severe disease: a study of the EULAR Scleroderma Trial and Research group». J Urol 188 (2): 549-50.
- University of Maryland Medical Center. «Scleroderma».
- The Scleroderma Foundation | Daniel Furst, M.D. (publicado originalmente en "Scleroderma Voice," 2002 #4)
Enlaces externos
- Página MedlinePlus (servicio de la Biblioteca Nacional de Medicina de los Estados Unidos y de los Institutos Nacionales de Salud) sobre la esclerodermia
| Control de autoridades |
|
|---|
 Datos: Q5340515
Datos: Q5340515 Multimedia: Systemic sclerosis / Q5340515
Multimedia: Systemic sclerosis / Q5340515