Metabolismo de fármacos
El metabolismo de fármacos es la descomposición metabólica de sustancias farmacéuticas o xenobióticos por parte de organismos vivos, generalmente a través de sistemas enzimáticos especializados. De forma más general, el metabolismo xenobiótico es el conjunto de vías metabólicas que modifican la estructura química de los xenobióticos, que son compuestos extraños a la bioquímica normal de un organismo, como cualquier medicamento o veneno.

Las reacciones de estas vías son de gran interés para la farmacología y la medicina. Por ejemplo, la tasa de metabolismo determina la duración e intensidad de la acción farmacológica de un medicamento. También afecta a la resistencia a múltiples fármacos en enfermedades infecciosas y a tratamientos de quimioterapia. Además, las acciones de algunos fármacos como sustratos o inhibidores de enzimas involucradas en el metabolismo de xenobióticos son una causa común de interacciones farmacológicas peligrosas.
También son importantes en ciencias ambientales, ya que el metabolismo xenobiótico de los microorganismos determina si un contaminante se degradará durante la biorremediación o si persistirá en el medio ambiente. Las enzimas del metabolismo de los xenobióticos, particularmente las glutatión S-transferasas (GST), también son importantes en la agricultura, ya que pueden producir resistencia a los pesticidas y herbicidas.
Barreras de permeabilidad y desintoxicación
Los compuestos exactos a los que está expuesto un organismo son en gran medida impredecibles y pueden diferir con el tiempo.[1] Para abordar este problema, las soluciones que se han dado con el tiempo han evolucionado en una combinación planificada de barreras físicas y sistemas enzimáticos de baja especificidad.
Todos los organismos usan membranas celulares como barreras de permeabilidad hidrófobas para controlar el acceso a su entorno interno. Los compuestos polares no pueden difundirse a través de estas, y la absorción de las moléculas útiles está mediada por proteínas de transporte que seleccionan específicamente sustratos de la mezcla extracelular. Esta absorción selectiva significa que la mayoría de las moléculas hidrófilas no pueden entrar en las células, ya que no son reconocidas por ningún transportador específico.[2] Por el contrario, la difusión de compuestos hidrófobos a través de estas barreras no se puede controlar, y por tanto, los organismos no pueden ecxcluir los xenobióticos liposolubles utilizando barreras de membrana.
Sin embargo, la existencia de una barrera de permeabilidad significa que los organismos fueron capaces de desarrollar sistemas de desintoxicación que explotan la hidrofobicidad común a los xenobióticos permeables a la membrana. Por lo tanto, estos sistemas resuelven el problema de la especificidad poseyendo especificidades de sustrato tan amplias que metabolizan casi cualquier compuesto no polar.[1] Los metabolitos útiles se excluyen ya que son polares, y en general contienen uno o más grupos cargados.
La desintoxicación de los subproductos reactivos del metabolismo normal no puede ser lograda por los sistemas descritos anteriormente, porque estas especies se derivan de componentes celulares normales y generalmente comparten sus características polares. Sin embargo, dado que estos compuestos son pocos en número, las enzimas específicas pueden reconocerlos y eliminarlos. Ejemplos de estos sistemas específicos de desintoxicación son el sistema de glioxalasa, que elimina el aldehído metilglioxal reactivo,[3] y los diversos sistemas antioxidantes que eliminan especies reactivas de oxígeno.[4]
Fases de desintoxicación
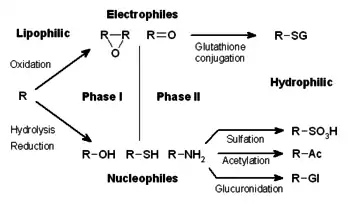
El metabolismo de los xenobióticos a menudo se divide en tres fases: modificación, conjugación y excreción. Estas reacciones actúan de manera coordinada para desintoxicar los xenobióticos y eliminarlos de las células.
Fase I: Modificación
En la fase I, una variedad de enzimas actúan para introducir grupos reactivos y polares en sus sustratos. Una de las modificaciones más comunes es la hidroxilación catalizada por el sistema oxidasa de función mixta dependiente del citocromo P450. Estos complejos enzimáticos actúan para incorporar un átomo de oxígeno en los hidrocarburos no activados, lo que puede resultar en la introducción de grupos hidroxilo o en la desalquilación N, O y S de sustratos.[5] El mecanismo de reacción de las oxidasas P-450 procede a través de la reducción del oxígeno unido al citocromo y la generación de una especie de oxiferrilo altamente reactiva, de acuerdo con el siguiente esquema: [6]
- O2 + NADPH + H+ + RH → NADP+ + H2O + ROH
Las reacciones de fase I (también llamadas reacciones no sintéticas) pueden ocurrir por oxidación, Redox, hidrólisis, ciclación, desciclación y adición de oxígeno o eliminación de hidrógeno, llevadas a cabo mediante oxidasas de función mixta, a menudo en el hígado. Estas reacciones oxidativas suelen involucrar una monooxigenasa de citocromo P450 (abreviada CYP), NADPH y oxígeno. Las clases de medicamentos farmacéuticos que utilizan este método para su metabolismo incluyen fenotiazinas, paracetamol y esteroides. Si los metabolitos de las reacciones de fase I son lo suficientemente polares, pueden excretarse fácilmente en este punto. Sin embargo, muchos productos de fase I no se eliminan rápidamente y sufren una reacción posterior en la que un sustratoendógeno se combina con el grupo funcional recién incorporado para formar un conjugado altamente polar.
Una oxidación común de fase I implica la conversión de un enlace C-H en uno C-OH. Esta reacción a veces convierte un compuesto farmacológicamente inactivo (un profármaco) en uno farmacológicamente activo. Del mismo modo, la Fase I puede convertir una molécula no tóxica en una venenosa (toxificación). La hidrólisis simple en el estómago es normalmente una reacción inocua, sin embargo, hay excepciones: por ejemplo, el metabolismo de fase I convierte el acetonitrilo en HOCH2CN, que se disocia rápidamente en formaldehído y cianuro de hidrógeno.[7]
El metabolismo de fase I de los candidatos a medicamentos se puede simular en un laboratorio utilizando catalizadores no enzimáticos.[8] Este ejemplo de una reacción biomimética tiende a dar productos que a menudo contienen los metabolitos de Fase I. Por ejemplo, el metabolito principal de la trimebutina, la desmetiltrimebutina (nor-trimebutina), se puede producir de manera eficiente mediante la oxidación in vitro del fármaco disponible comercialmente. La hidroxilación de un grupo N-metilo conduce a la expulsión de una molécula de formaldehído, mientras que la oxidación de los grupos O-metilo tiene lugar en menor medida.
Oxidación
- Monooxigenasa de Citocromo P450
- Monooxigenasa que contiene flavina
- Alcohol deshidrogenasa y Aldehído deshidrogenasa
- Monoamino oxidasa
- Co-oxidación por Peroxidasas
Reducción
- Citocromo P450 reductasa
La citocromo P450 reductasa, también conocida como NADPH:ferrihemoproteína oxidorreductasa, NADPH:hemoproteína oxidorreductasa, NADPH:P450 oxidorreductasa, P450 reductasa, POR, RCP, CYPOR, es una enzima unida a membrana necesaria para la transferencia de electrones al citocromo P450 en el microsoma de la célula eucariota desde una enzima que contiene FAD y FMN NADPH:citocromo P450 reductasa. El esquema general del flujo de electrones en el sistema POR/P450 es:
- NADPH → FAD → FMN → P450 → O2
- Citocromo P450 reducido (ferroso)
Durante las reacciones de reducción, una sustancia química puede entrar en ciclos inútiles, en el que gana un electrón de radicales libres, y luego lo pierde rápidamente por oxígeno (para formar un anión superóxido).
Fase II: Conjugación
En reacciones posteriores de fase II, estos metabolitos xenobióticos activados son conjugados con especies cargadas como glutatión (GSH), sulfato, glicina o ácido glucurónico. Los lugares donde se producen reacciones de conjugación en los medicamentos incluyen los grupos carboxilo (-COOH), hidroxilo (-OH), amino (NH2) y tiol (-SH). Los productos de las reacciones de conjugación aumentan el peso molecular y tienden a ser menos activos que sus sustratos, a diferencia de las reacciones de fase I que a menudo producen metabolitos activos. La adición de grandes grupos aniónicos (como GSH) desintoxica los electrófilos reactivos y produce más metabolitos polares que no pueden difundirse a través de las membranas y, por lo tanto, pueden ser transportados activamente.
Estas reacciones son catalizadas por un gran grupo de transferasas de amplia especificidad, que, en combinación, pueden metabolizar casi cualquier compuesto hidrófobo que contenga grupos nucleofílicos o electrofílicos.[1] Una de las clases más importantes de este grupo es la de la glutatión S-transferasas (GST).
| Mecanismo | Enzima involucrada | Cofactor | Ubicación | Referencia |
|---|---|---|---|---|
| Metilación | Metiltransferasa | S-adenosil metionina | Hígado, riñón, pulmón, SNC | [9] |
| Sulfatación | Sulfotransferasas | 3'-Fosfoadenosina-5'-fosfosulfato | Hígado, riñón, intestino | [9] |
| Acetilación |
|
Coenzima acetil A | Hígado, pulmón, bazo, mucosa gástrica, glóbulos rojos, linfocitos | [9] |
| Glucuronidación | Glucuroniltransferasas | Ácido glucurónico difosfato de uridina | Hígado, riñón, intestino, pulmón, piel, próstata, cerebro | [9] |
| Conjugación de Glutatión | Glutatión S-transferasas | Glutatión | Hígado, riñón | [9] |
| Conjugación de glicina | Proceso en dos pasos:
|
Glicina | Hígado, riñón | [10] |
Fase III: Modificaciones adicionales y excreción
Tras las reacciones de fase II, los conjugados xenobióticos pueden metabolizarse aún más. Un ejemplo común es el procesamiento de conjugados de glutatión a conjugados de acetilcisteína (ácido mercapúrico).[11] Como último paso, el residuo de cisteína en el conjugado es acetilado.
Los conjugados y sus metabolitos se pueden excretar de las células en la fase III de su metabolismo, con los grupos aniónicos actuando como marcadores de afinidad para una variedad de transportadores de membrana de la familia de la Glucoproteína-P.[12] Estas proteínas son miembros de la familia de los transportadores ABC y pueden catalizar el transporte dependiente del ATP de una gran variedad de aniones hidrófobos,[13] y así actuar para eliminar los productos de fase II al medio extracelular, donde pueden ser metabolizados o excretados aún más.[14]
Toxinas endógenas
La desintoxicación de metabolitos reactivos endógenos como peróxidos y aldehídos reactivos a menudo no puede ser lograda por el sistema descrito anteriormente. Este se debe a que estas especies se derivan de constituyentes celulares normales y generalmente comparten sus características polares. Sin embargo, dado que estos compuestos son pocos en número, es posible que los sistemas enzimáticos utilicen reconocimiento molecular específico para reconocerlos y eliminarlos. La similitud de estas moléculas con los metabolitos útiles significa que generalmente se requieren diferentes enzimas de desintoxicación para el metabolismo de cada grupo de toxinas endógenas. Algunos ejemplos de estos sistemas de desintoxicación específicos son el sistema de glioxalasa, que actúa para deshacerse del aldehído reactivo metilglioxal, y los diversos sistemas antioxidantes que eliminan especies reactivas de oxígeno.
Ubicaciones
Cuantitativamente, el retículo endoplásmico liso de la célula hepática es el principal órgano en el metabolismo de medicamentos, aunque cada tejido biológico tiene cierta capacidad para metabolizar los fármacos. El hígado es de tal importancia en este proceso debido a que es un órgano grande, que es el primer órgano perfundido por sustancias químicas absorbidas en el intestino y porque hay concentraciones muy altas de la mayoría de los sistemas enzimáticos metabolizadores de fármacos con respecto a otros órganos. Si un medicamento se toma en el tracto gastrointestinal, donde entra en circulación hepática a través de la vena porta, se metaboliza bien y se dice que muestra un efecto de primer paso.
Otros lugares donde tiene lugar el metabolismo de medicamentos son las células epiteliales del tracto gastrointestinal, los pulmones, los riñones y la piel. Estos suelen ser responsables de las reacciones de toxicidad localizadas.
Factores que afectan al metabolismo de fármacos
La duración e intensidad de la acción farmacológica de la mayoría de los medicamentos lipofílicos está determinada por la velocidad en que se metabolizan en productos inactivos. El Citocromo P450 es la vía más importante a este respecto.
Por lo general, cualquier cosa que aumente la tasa de metabolismo (por ejemplo, en la inducción enzimática) de un metabolito farmacológicamente activo, disminuirá la duración e intensidad de la acción del medicamento. Lo contrario también puede ser cierto (por ejemplo, en la inhibición enzimática); sin embargo, en los casos en que una enzima es responsable de metabolizar un profármaco en un medicamento, la inducción enzimática puede acelerar esta conversión y aumentar los niveles de fármaco, causando potencialmente toxicidad.
La dosis, la frecuencia, la vía de administración, la distribución tisular y la unión a proteínas del medicamento afectan a su metabolismo.
Existen factores fisiológicos y patológicos que también pueden afectar al metabolismo de los medicamentos. Los factores fisiológicos pueden ser la edad, el sexo, la farmacogenómica, la circulación enterohepática, la nutrición o la flora intestinal. Los factores patológicos pueden ser las enfermedades del hígado, del pulmón o del corazón. Además, por norma general los fármacos se metabolizan más lentamente en fetos, neonatos y en humanos y animales ancianos.
La variación genética (polimorfismo) explica parte de la variabilidad en el efecto de los medicamentos. Con las N-acetiltransferasas (involucradas en reacciones de Fase II), la variación individual crea un grupo de personas que acetilan lentamente (acetiladores lentos) y aquellas que acetilan rápidamente. En Canadá se calculó que aproximadamente la mitad de la población pertenece a cada uno de estos grupos. Esta variación puede tener consecuencias dramáticas, ya que los acetiladores lentos son más propensos a la toxicidad por sobredosis.
Las enzimas de monooxigenasa del citocromo P450 también pueden variar entre los individuos, con deficiencias que ocurren a entre el 1% y el 30% de la población, dependiendo de su origen étnico.
Los métodos de modelado y simulación In silico permiten predecir el metabolismo de los medicamentos en poblaciones de pacientes virtuales antes de realizar estudios clínicos en seres humanos.[15] Esto puede ser útil para identificar a las personas con mayor riesgo de reacciones adversas.
Historia
Los estudios sobre cómo las personas transforman las sustancias que ingieren comenzaron a mediados del siglo XIX, cuando los químicos descubriendo que las sustancias orgánicas como el benzaldehído podrían oxidarse y conjugarse con aminoácidos en el cuerpo humano.[16] Durante el resto del siglo XIX, se descubrieron otras reacciones básicas de desintoxicación, como la metilación, la acetilación y la sulfonación.
A principios del siglo XX, el trabajo pasó a la investigación de las enzimas y de las vías responsables de la producción de estos metabolitos. Este campo se definió como un área de estudio separada con la publicación del libro Detoxication mechanisms (Mecanismos de desintoxicación) por Richard Williams en 1947.[17] Esta investigación bioquímica moderna resultó en la identificación de las glutatión S-transferasas (GST) en 1961,[18] seguida del descubrimiento del citocromo P450s en 1962,[19] y la investigación de su papel en el metabolismo xenobiótico en 1963.[20][21]
Véase también
- Antioxidante
- Biodegradación
- Biorremediación
- Biodegradación microbiana
Referencias
- Jakoby WB, Ziegler DM (Diciembre 1990). «The enzymes of detoxication». J. Biol. Chem. 265 (34): 20715-8. PMID 2249981. doi:10.1016/S0021-9258(17)45272-0. Archivado desde el original el 21 de junio de 2009. Consultado el 20 de febrero de 2022.
- Mizuno N, Niwa T, Yotsumoto Y, Sugiyama Y (Septiembre 2003). «Impact of drug transporters studies on drug discovery and development». Pharmacol. Rev. 55 (3): 425-61. PMID 12869659. S2CID 724685. doi:10.1124/pr.55.3.1.
- Thornalley PJ (Julio 1990). «The glyoxalase system: new developments towards functional characterization of a metabolic pathway fundamental to biological life». Biochem. J. 269 (1): 1-11. PMC 1131522. PMID 2198020. doi:10.1042/bj2690001.
- Sies H (Marzo 1997). «Oxidative stress: oxidants and antioxidants». Exp. Physiol. 82 (2): 291-5. PMID 9129943. doi:10.1113/expphysiol.1997.sp004024.
- Guengerich FP (Junio 2001). «Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity». Chem. Res. Toxicol. 14 (6): 611-50. PMID 11409933. doi:10.1021/tx0002583.
- Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG (Marzo 2000). «The catalytic pathway of cytochrome p450cam at atomic resolution». Science 287 (5458): 1615-22. Bibcode:2000Sci...287.1615S. PMID 10698731. doi:10.1126/science.287.5458.1615.
- «Acetonitrile (EHC 154, 1993)». www.inchem.org.
- Akagah B, Lormier AT, Fournet A, Figadère B (Diciembre 2008). «Oxidation of antiparasitic 2-substituted quinolines using metalloporphyrin catalysts: scale-up of a biomimetic reaction for metabolite production of drug candidates». Org. Biomol. Chem. 6 (24): 4494-7. PMID 19039354. doi:10.1039/b815963g.
- Liston HL, Markowitz JS, DeVane CL (Octubre 2001). «Drug glucuronidation in clinical psychopharmacology». J Clin Psychopharmacol 21 (5): 500-15. PMID 11593076. S2CID 6068811. doi:10.1097/00004714-200110000-00008.
- Badenhorst CP, van der Sluis R, Erasmus E, van Dijk AA (Septiembre 2013). «Glycine conjugation: importance in metabolism, the role of glycine N-acyltransferase, and factors that influence interindividual variation». Expert Opinion on Drug Metabolism & Toxicology 9 (9): 1139-1153. PMID 23650932. S2CID 23738007. doi:10.1517/17425255.2013.796929.
- Boyland E, Chasseaud LF (1969). «The role of glutathione and glutathione S-transferases in mercapturic acid biosynthesis». Adv. Enzymol. Relat. Areas Mol. Biol. Advances in Enzymology – and Related Areas of Molecular Biology 32: 173-219. ISBN 9780470122778. PMID 4892500. doi:10.1002/9780470122778.ch5.
- Homolya L, Váradi A, Sarkadi B (2003). «Multidrug resistance-associated proteins: Export pumps for conjugates with glutathione, glucuronate or sulfate». BioFactors 17 (1–4): 103-14. PMID 12897433. S2CID 7744924. doi:10.1002/biof.5520170111.
- König J, Nies AT, Cui Y, Leier I, Keppler D (Diciembre 1999). «Conjugate export pumps of the multidrug resistance protein (MRP) family: localization, substrate specificity, and MRP2-mediated drug resistance». Biochim. Biophys. Acta 1461 (2): 377-94. PMID 10581368. doi:10.1016/S0005-2736(99)00169-8.
- Commandeur JN, Stijntjes GJ, Vermeulen NP (Junio 1995). «Enzymes and transport systems involved in the formation and disposition of glutathione S-conjugates. Role in bioactivation and detoxication mechanisms of xenobiotics». Pharmacol. Rev. 47 (2): 271-330. PMID 7568330.
- Rostami-Hodjegan A, Tucker GT (Febrero 2007). «Simulation and prediction of in vivo drug metabolism in human populations from in vitro data». Nat Rev Drug Discov 6 (2): 140-8. PMID 17268485. S2CID 205476485. doi:10.1038/nrd2173.
- Murphy PJ (Junio 2001). «Metabolismo xenobiótico: una mirada del pasado al futuro». Drug Metab. Dispos. 29 (6): 779-80. PMID 11353742.
- Neuberger A, Smith RL (1983). «Richard Tecwyn Williams: the man, his work, his impact». Drug Metab. Rev. 14 (3): 559-607. PMID 6347595. doi:10.3109/03602538308991399.
- Booth J, Boyland E, Sims P (Junio 1961). «An enzyme from rat liver catalysing conjugations with glutathione». Biochem. J. 79 (3): 516-24. PMC 1205680. PMID 16748905. doi:10.1042/bj0790516.
- Omura T, Sato R (Abril 1962). «A new cytochrome in liver microsomes». J. Biol. Chem. 237 (4): 1375-6. PMID 14482007. doi:10.1016/S0021-9258(18)60338-2. Archivado desde el original el 21 de junio de 2009. Consultado el 20 de febrero de 2022.
- Estabrook RW (Diciembre 2003). «A passion for P450s (remembrances of the early history of research on cytochrome P450)». Drug Metab. Dispos. 31 (12): 1461-73. PMID 14625342. doi:10.1124/dmd.31.12.1461.
- Estabrook RW, Cooper DY, Rosenthal O (1963). «The light reversible carbon monoxide inhibition of steroid C-21 hydroxylase system in adrenal cortex». Biochem Z 338: 741-55. PMID 14087340.
Bibliografía
- Parvez H, Reiss C (2001). Elsevier, ed. Molecular Responses to Xenobiotics. ISBN 0-345-42277-5.
- Ioannides C (2001). John Wiley and Sons, ed. Enzyme Systems That Metabolise Drugs and Other Xenobiotics. ISBN 0-471-89466-4.
- Richardson M (1996). Taylor & Francis Ltd, ed. Environmental Xenobiotics. ISBN 0-7484-0399-X.
- Ioannides C (1996). CRC Press Inc, ed. Cytochromes P450: Metabolic and Toxicological Aspects. ISBN 0-8493-9224-1.
- Awasthi YC (2006). CRC Press Inc, ed. Toxicology of Glutathionine S-transferses. ISBN 0-8493-2983-3.
| Control de autoridades |
|
|---|
 Datos: Q1124842
Datos: Q1124842 Multimedia: Drug metabolism / Q1124842
Multimedia: Drug metabolism / Q1124842