Neurotoxina
Las neurotoxinas son una clase extensa de sustancias químicas exógenas neurológicamente dañinas[3] que pueden causar efectos adversos en la función tanto del tejido nervioso en desarrollo como en el maduro.[4] El término neurotoxina deriva del griego antiguo νευρών (nevron) “nervio” y τοξικόν (toxikon) “toxina”. También puede ser usado para clasificar compuestos endógenos que cuando están presentes en concentraciones anormales pueden convertirse en neurológicamente tóxicos.[3] Aunque las neurotoxinas suelen ser neurológicamente destructivas, su habilidad para tener como objetivo específico los componentes neurales es importante en el estudio de los sistemas nerviosos.[5] Ejemplos comunes de neurotoxinas incluyen plomo,[6] etanol,[7] glutamato,[8] óxido nítrico (NO),[9] toxina botulínica,[10] toxina tetánica[11] y tetrodotoxina.[5]

La actividad de las neurotoxinas puede ser caracterizada por la habilidad de inhibir el control neuronal sobre las concentraciones de iones a través de la membrana celular[5] o la comunicación entre las neuronas a través de la sinapsis.[12]
Las patologías locales de la exposición a neurotoxinas suelen incluir excitotoxicidad o apoptosis neuronal,[13] pero también puede incluir daño de las células gliales.Las manifestaciones macroscópicas de exposición a neurotoxinas pueden incluir daño extendido al sistema nervioso central como retraso mental,[4] deficiencia de memoria persistente,[14] epilepsia y demencia.[15] Adicionalmente, es común el daño del sistema nervioso periférico mediado por neurotoxinas como la neuropatía o la miopatía. Han sido demostrados tratamientos de ayuda frente a los daños causados por neurotoxinas tales como la administración de antioxidantes,[7] antitoxinas[16] y etanol.[17]
Antecedentes
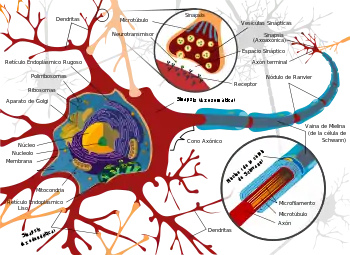
La exposición a neurotoxinas en la sociedad no es nueva, pues las civilizaciones han estado expuestas a compuestos neurológicamente destructivos durante miles de años. Un ejemplo notable es la significativa exposición a plomo durante el Imperio Romano resultante del desarrollo de la extensa red de alcantarillado y del hábito de hervir vino avinagrado en cazuelas de plomo para endulzarlo, proceso que generaba acetato de plomo conocido como “azúcar de plomo”.[18] En parte, las neurotoxinas han sido parte de la historia de la humanidad debido a la frágil y susceptible naturaleza del sistema nervioso, creando una alta tendencia a la disrupción.
El tejido nervioso presente en el cerebro, en la médula espinal y en la periferia comprende un extraordinario y complejo sistema biológico que define en gran medida muchos de los rasgos únicos de los individuos. Al igual que cualquier sistema muy complejo, incluso pequeñas perturbaciones en su entorno pueden provocar alteraciones funcionales significativas. Entre las propiedades que conducen a la susceptibilidad de los tejidos nerviosos se incluyen: la presencia de una gran superficie de neuronas, un alto contenido de lípidos que retiene toxinas lipofílicas, un alto flujo de sangre hacia el cerebro lo que induce una mayor exposición efectiva a la toxina, y la persistencia de las neuronas a través de la vida del individuo, lo que agrava los daños y perjuicios.[19] Como resultado, el sistema nervioso tiene numerosos mecanismos diseñados para protegerlo frente a ataques internos y externos, incluyendo la barrera hematoencefálica.
La barrera hematoencefálica es un ejemplo crítico de protección el cual previene que toxinas y otros compuestos adversos alcancen el cerebro.[20] Como el cerebro requiere la entrada de nutrientes y la salida de residuos, es prefundido por el flujo sanguíneo. La sangre puede transportar un gran número de tóxicos ingeridos, que inducirían una significativa muerte de neuronas si ellas alcanzaran el tejido nervioso. Por consiguiente, las células protectoras denominadas astrocitos rodean los capilares del cerebro y absorben los nutrientes de la sangre para posteriormente transportarlos a las neuronas, aislando con eficiencia al cerebro de un gran número de potenciales agentes químicos nocivos.[20]
Esta barrera crea una capa hidrófoba alrededor de los capilares del cerebro inhibiendo el trasporte de compuestos grandes o hidrófilos. Además de la barrera hematoencefálica, el plexo coroideo proporciona una capa de protección contra la absorción de toxinas en el cerebro. Los plexos coroideos son capas de tejido vascularizado que se encuentran en los ventrículos tercero, cuarto, ventrículos laterales del cerebro, que a través de la función de sus células ependimarias, son responsables de la síntesis de líquido cefalorraquídeo.[21] Es importante destacar que, a través del paso selectivo de iones y nutrientes y la captación de metales pesados tales como el plomo, los plexos coroideos mantienen un entorno estrictamente regulado que contiene al cerebro y la médula espinar debido a que están rodeados de líquido cefalorraquídeo.[20][21]
Al ser hidrofóbicos y pequeños, o inhibir la función de los astrocitos, algunos compuestos entre los que se incluyen ciertas neurotoxinas son capaces de penetrar en el cerebro e inducir daños significativos. Actualmente, a los científicos y los médicos se les ha presentado el reto de identificar y tratar a las neurotoxinas, lo que ha dado lugar a un creciente interés en la investigación de la Neurotoxicología y los estudios clínicos.[22] La neurotoxicología clínica es en gran parte un campo floreciente, se han realizado extensos avances en la identificación de muchas neurotoxinas ambientales que han conducido a la clasificación de 750 a 1000 compuestos conocidos como potencialmente neurotóxicos.[19] Debido a la gran importancia de encontrar de neurotoxinas en ambientes comunes, diversos protocolos específicos han sido desarrollados por la Agencia de Protección Ambiental de Estados Unidos (EPA) para probar y determinar los efectos neurotóxicos de los compuestos (USEPA 1998). Además, el uso de sistemas in vitro ha aumentado, ya que proporcionan mejoras significativas frente a los sistemas in vivo del pasado. Ejemplos de mejoras incluyen ambientes uniformes manejables, y la eliminación de la contaminación de los efectos del metabolismo sistémico.[22] Sin embargo, los sistemas in vitro han presentado problemas, ya que ha sido difícil replicar adecuadamente las complejidades del sistema nervioso, tales como las interacciones entre los astrocitos y las neuronas de apoyo en la creación de la barrera hematoencefálica.[23] Para complicar aun más el proceso de la determinación de neurotoxinas cuando se prueba in vitro, podría no distinguirse bien la citotoxicidad o la neurotoxicidad, ya que exponer a las neuronas directamente a las neurotoxinas podría no ser posible in vivo, y sí que lo es in vitro. Además, la respuesta de las células a productos químicos no puede transmitir con precisión una distinción entre neurotoxinas y citotoxinas, ya que síntomas como el estrés oxidativo o las modificaciones del citoesqueleto podrían ocurrir en respuesta a ambas.[24]
En un esfuerzo por abordar esta complicación, recientemente se han propuesto las prolongaciones de las neuronas (ya sean las dendritas o el axón en respuesta a compuestos aplicados como una distinción más precisa entre las verdaderas neurotoxinas y citotoxinas en un entorno de pruebas in vitro. Debido a las imprecisiones significativas asociadas este proceso ha tardado mucho en ganar apoyo.[25] Además, los mecanismos bioquímicos se han vuelto más ampliamente utilizados en las pruebas de neurotoxinas, de tal manera que se puede hacer un cribado de los compuestos químicos para estudiar su capacidad de inducir una interferencia en un mecanismo celular, como la capacidad de los organofosforados (que incluyen el DDT y gas sarín) de inhibir a la acetilcolinesterasa.[26] Aunque los métodos de determinación de la neurotoxicidad todavía requieren un importante desarrollo, la identificación de compuestos nocivos y síntomas de exposición a las toxinas han experimentado una mejoría significativa.
Mecanismos de actividad
Como las neurotoxinas son compuestos que afectan adversamente al sistema nervioso, muchos de los mecanismos a través de los cuales funcionan tienen lugar a través de la inhibición de los procesos celulares neuronales. Estos procesos inhibidos pueden ir desde los mecanismos de despolarización de membrana hasta la comunicación entre neuronas. Mediante la inhibición de la capacidad de las neuronas para llevar a cabo sus funciones intracelulares esperadas, o pasar una señal a una neurona vecina, las neurotoxinas pueden inducir la detención del sistema nervioso sistémico como en el caso de la toxina botulínica,[12] o incluso la muerte del tejido nervioso.[27] El tiempo requerido para la aparición de los síntomas tras la exposición a la neurotoxina puede variar entre las diferentes toxinas, siendo del orden de horas para la toxina botulínica[16] y de años para el plomo.[28]
| Clasificación de las neurotoxinas | Neurotoxinas |
|---|---|
| Inhibidores de los canales de Na | Tetrodotoxina[5] |
| Inhibidores de los canales de K | Tetraetilamonio[29] |
| Inhibidores de los canales de Cl | Clorotoxina[30] |
| Inhibidores de los canales de Ca | Conotoxina[31] |
| Inhibidores de la liberación de vesículas sinápticas | Toxina botulínica,[32] Toxina tetánica[33] |
| Inhibidores de receptores | Bungarotoxina,[34] Curare[35] |
| Inhibidores de la barrera hematoencefálica | Aluminio,[36] Mercurio[37] |
| Interferencia con el citoesqueleto | Arsénico,[38] Amoníaco[39] |
| Citotoxicidad mediada por Ca | Plomo[40] |
| Efectos múltiples | Etanol,[41][42] n-Hexano[43][44][45][46] |
| Fuentes endógenas de neurotoxinas | Óxido nítrico,[47] Glutamato[8] |
Tetradotoxina

La tetradotoxina (TTX) es un veneno producido por organismos que pertenecen al orden Teradontidae, que incluye al pez globo, el pez luna y el pez erizo.[48] En el pez globo, que es un manjar común especialmente en Japón, la TTX está presente en el hígado, las gónadas, los ovarios, los intestinos y la piel.[5][49] La TTX puede ser mortal si se consume, y se ha convertido en una forma común de intoxicación en muchos países. Los síntomas comunes del consumo de TTX incluyen parestesias (a menudo restringidas a la boca y las extremidades), debilidad muscular, náuseas y vómitos[8] que a menudo se manifiestan dentro de los 30 minutos tras la ingestión.[50] El mecanismo principal por el cual la TTX es tóxica es mediante la inhibición de la función del canal de sodio, que reduce la capacidad funcional de la comunicación neuronal. Esta inhibición afecta en gran medida a un subconjunto de canales de sodio conocidos como sensibles a la TTX (TTX-s), que también son en gran parte responsables de la corriente de sodio que impulsa la fase de despolarización de los potenciales de acción neuronales.[5]
La TTX-resistente (TTX-R) es otra forma de canal de sodio que tiene una sensibilidad limitada a la TTX, y que se encuentra en gran parte en los axones de pequeño diámetro, tales como los que se encuentran en las neuronas de nocicepción.[5] Cuando niveles significativos de TTX son ingeridos, se enlazará a los canales de sodio en las neuronas y se reducirá así la permeabilidad de la membrana al sodio. Esto se traduce en un aumento del umbral efectivo de señales excitatorias requeridas para inducir un potencial de acción de una neurona postsináptica. El efecto de este aumento del umbral de señalización provoca una excitabilidad reducida de las neuronas postsinápticas, y la subsiguiente pérdida de la función motora y sensorial que puede dar lugar a parálisis y muerte. A pesar de que la asistencia respiratoria puede aumentar las posibilidades de supervivencia después de la exposición a TTX, actualmente no hay antitoxina. El uso de la neostigmina (inhibidor de la acetilcolinesterasa) o de atropina (antagonista de acetilcolina)(que inhibe la actividad parasimpática), puede aumentar la actividad del sistema nervioso simpático suficientemente para mejorar la probabilidad de supervivencia después de la exposición a TTX.[48]
Tetraetilamonio
El tetraetilamonio (TEA) es un compuesto que, al igual que otras neurotoxinas, fue identificado por primera vez a través de sus efectos perjudiciales para el sistema nervioso y se ha demostrado que tiene la capacidad de inhibir la función de los nervios motores y en consecuencia la contracción de la musculatura, de manera similar al curare.[51] Además, a través de la administración crónica de TEA se induciría la atrofia muscular.[51] Más tarde se determinó que el TEA funciona in vivo primariamente a través de su habilidad para inhibir tanto los canales de potasio responsables de la repolarización de membrana vista en un potencial de acción, como algunas poblaciones de canales de potasio calcio-dependientes.[29] Es esta capacidad para inhibir el flujo de potasio en las neuronas lo que ha hecho del TEA una de las herramientas más importantes en neurociencia. Se ha planteado la hipótesis de que la capacidad del TEA para inhibir los canales de potasio se deriva de su estructura de llenado de espacio similar a los iones de potasio.[51] Lo que hace al TEA muy útil para los neurólogos es su capacidad específica para eliminar la actividad de los canales de potasio, permitiendo así el estudio de la contribución en la respuesta de las neuronas de otros canales iónicos como los canales de sodio dependientes de voltaje.[52] Además de sus muchos usos en la investigación en neurociencias, el TEA ha demostrado actuar como un tratamiento eficaz de la enfermedad de Parkinson a través de su capacidad de limitar la progresión de la enfermedad.[29]
Clorotoxina
La clorotoxina (CLTX) es el compuesto activo que se encuentra en el veneno del escorpión, y cuya toxicidad es debida principalmente a su capacidad para inhibir la conductancia de los canales de cloruro.[30] La ingestión de volúmenes letales de CLTX resulta en parálisis a través de la interrupción de este canal iónico. Similar a la toxina botulínica, la CLTX ha demostrado poseer un valor terapéutico significativo. La evidencia es que la CLTX puede inhibir la capacidad de los gliomas de infiltrarse en el tejido nervioso sano del cerebro, lo que reduce significativamente el potencial daño invasivo causado por tumores.[53][54]
Conotoxina
Las conotoxinas representan un grupo de venenos producidos por los caracoles cono marinos (familia Conidae), y son capaces de inhibir la actividad de un gran número de canales de iones tales como calcio, sodio o potasio.[55][56] En muchos casos, las toxinas liberadas por los diferentes tipos de caracoles cono incluyen una gama de diferentes tipos de conotoxinas, que pueden ser específicas para diferentes canales de iones creando así un veneno capaz de generar una interrupción generalizada de la función nerviosa.[55] Una de las formas de la conotoxina, la ω-conotoxina (ω-CgTx) es altamente específica para los canales de Ca y ha demostrado ser útil en el aislamiento de ellos a partir de un sistema.[57] Como el flujo de calcio es necesario para la buena excitabilidad de la célula, cualquier importante inhibición podría evitar una gran cantidad de función. Significativamente la ω-CgTx es capaz de unirse a largo plazo e inhibir los canales de calcio dependientes de voltajes localizados en las membranas de las neuronas, pero no a los de las células musculares.[31]
Toxina botulínica

La toxina botulínica (BTX) es un grupo de neurotoxinas que consta de ocho compuestos distintos, a los que se refiere como BTX- A, B, C, D, E, F, G, H, que son producidos par la bacteria Clostridium botulinum y que dan lugar a la parálisis muscular.[58] Una característica particular única de la toxina botulínica es su uso terapéutico relativamente común en el tratamiento de la distonía y trastornos de espasticidad,[58] así como en la inducción de atrofia muscular[10] a pesar de ser la sustancia más venenosa conocida. El funcionamiento de la BTX consiste en inhibir la liberación de acetilcolina (ACh) en la unión neuromuscular a través de la degradación de las proteínas SNARE requeridas para la fusión de las vesículas de Ach con la membrana.[32] Como la toxina es biológicamente muy activa, una dosis estimada de 1 μg/kg de peso corporal es suficiente para inducir una insuficiencia en el volumen de la ventilación pulmonar y causar la resultante muerte por asfixia.[12] Debido a su alta toxicidad, la búsqueda de antitoxinas BTX ha sido un área muy activa de investigación. Se ha demostrado que la capsaicina (compuesto activo responsable del sabor picante de los chiles) puede unirse al receptor TRPV1 expresando en las neuronas colinérgicas e inhibir los efectos tóxicos de la toxina botulínica.[16]
Toxina tetánica
La neurotoxina tetánica (TeNT) es un compuesto que reduce funcionalmente las trasmisiones inhibidoras en el sistema nervioso lo cual resulta en tetania muscular. La TeNT es similar a la BTX, tanto en su estructura como origen. Ambas pertenecen a la misma categoría de las neurotoxinas clostridiales.[11] Al igual que la BTX, la TeNT inhibe la comunicación entre las neuronas a través de la liberación de vesículas de neurotransmisores.[33] Una diferencia notable entre los dos compuestos es que mientras la BTX inhibe las contracciones musculares, la TeNT las induce. Aunque ambas toxinas inhiben la liberación de vesículas en las sinapsis neuronales, la manifestación es diferente en cada caso, puesto que la acción de la BTX es principalmente en el sistema nervioso periférico (SNP), frente a la TeNT cuya acción es en gran parte activa en el sistema nervioso central (SNC).[59] Esto es un resultado de la migración del TeNT a través de las neuronas motoras a las neuronas inhibidoras de la médula espinal después de entrar a través de endocitosis,[60] lo cual se traduce en una pérdida de la función de las neuronas inhibitorias en el SNC resultante en las contracciones musculares sistémicas. Al igual que en el pronóstico de una dosis letal de la toxina botulínica, la TeNT conduce a la parálisis y la posterior asfixia.[60]
Aluminio
La neurotoxicidad del aluminio tiene lugar tras la entrada en el sistema circulatorio, donde puede migrar al cerebro e inhibir algunas de las funciones cruciales de la barrera hematoencefálica.[36] Una pérdida de la función de la barrera hematoencefálica puede producir un daño significativo en las neuronas del SNC, debido a que se perdería la función de la barrera hematoencefálica de proteger al cerebro de otras toxinas que se encuentran en la sangre. Aunque el metal es conocido por ser neurotóxico, los efectos se restringen generalmente a los pacientes incapaces de eliminar el exceso de iones en sangre, tales como los que experimentan insuficiencia renal.[61] Los pacientes que experimenta toxicidad del aluminio pueden presentar síntomas tales como problemas de aprendizaje y la reducción de la coordinación motora.[62] Además, se ha demostrado que la enfermedad de Alzheimer se correlaciona con el aumento de los niveles sistémicos de aluminio con la edad, por lo que podría tratarse de un compuesto neurotóxico causante de la enfermedad.[63]
Mercurio
El mercurio es capaz de inducir un daño en el SNC mediante la migración al cerebro, cruzando la barrera hematoencefálica y favoreciendo el envenenamiento por mercurio.[37] El mercurio está presente en un gran número de compuestos, aunque el metilmercurio (MeHg +), el dimetilmercurio y el dietilmercurio se consideran las únicas formas significativamente neurotóxicas. El dimetilmercurio y el dietilmercurio se consideran unas de las neurotoxinas más potentes jamás descubiertas.[37] El MeHg + se adquiere normalmente mediante el consumo de mariscos, ya que tiende a concentrándose en los organismos de altos estratos en la cadena alimenticia.[64] Se sabe que los iones de mercurio inhiben el transporte de aminoácidos (AA) y glutamato (Glu), lo que puede conducir a efectos de exocitotoxicidad.[65]
Bungarotoxina
Bungarotoxina es un compuesto conocido con la interacción con los receptores nicotínicos de acetilcolina (nAChR), que constituyen una familia de canales iónicos cuya actividad se activa por la unión del neurotransmisor.[66] La bungarotoxina se produce de diferentes formas, aunque una de las más utilizadas es la forma de una larga cadena alfa, α - bungarotoxina, que se aísla de la serpiente Bungarus fasciatus.[34] Aunque es extremadamente tóxico si se ingiere, α - bungarotoxina ha mostrado extensa utilidad en la neurociencia, ya que puede aislar con facilidad la nAChRs debido a su alta afinidad con los receptores.[34] Dado que hay múltiples formas de bungarotoxina, hay diferentes formas de nAChRs a los que se unirán, y α - bungarotoxina es particularmente específica para α7 - nAChR.[67] Este α7 - nAChR permite la afluencia de iones de calcio en las células, y por lo tanto cuando se bloquea por la ingestión de bungarotoxina producirá efectos perjudiciales, ya que la ACh de señalización será inhibida del mismo modo,[67] el uso de α - bungarotoxina puede ser muy útil en la neurociencia si se quiere bloquear el flujo de calcio con el fin de aislar los efectos sobre otros canales. Además, diferentes formas de bungarotoxina pueden ser útiles para el estudio de los nAChR inhibidos y su flujo de iones de calcio resultante en diferentes sistemas del cuerpo. Por ejemplo, α - bungarotoxina es específico para los nAChR que se sitúan en la musculatura y κ - bungarotoxina es específico para los nAChR situados en las neuronas.[68]
Anatoxina-a
| Video externo | ||
|---|---|---|
|
University of Nottingham | ||
Atención: este archivo está alojado en un sitio externo, fuera del control de la Fundación Wikimedia. |

Los estudios sobre la anatoxina-a, también conocidos como "Very Fast Death Factor" (Factor de muerte muy rápida), comenzaron en el año 1961 tras la muerte de varias vacas que habían bebido agua de un lago que contenía una gran cantidad de algas en Saskatchewan, Canadá.[69][70] Se trataba de una cianotoxina producida por al menos cuatro géneros diferentes de cianobacterias, se había informado ya en Norte América, Europa, África, Asia y Nueva Zelanda.[71]
Los efectos tóxicos de la anatoxina-a progresan muy rápidamente, ya que actúan directamente sobre las células nerviosas (neuronas). Los síntomas progresivos debido a la exposición a esta toxina son: la pérdida de coordinación, temblores, convulsiones y una muerte rápida por parada respiratoria. Los tejidos nerviosos conectados con los músculos contienen un receptor denominado acetilcolina nicotínico. La estimulación de estos receptores provoca una contracción muscular. La anatoxina-a encaja con este receptor imitando al neurotransmisor natural normalmente utilizado. Una vez realizada la contracción muscular la anatoxina-a no permite que el músculo vuelva a su estado de reposo, ya que no se degrada con la colinesterasa (la cual realiza normalmente esta función). Como resultado las células del músculo permanecen constantemente contraídas, la comunicación entre el cerebro y los músculos se interrumpe y se detiene la respiración.[72][73]
Cuando se descubrió por primera vez, fue llamada "Very Fast Death Factor" (VFDF) porque al inyectarla en la cavidad torácica de los ratones producía temblores convulsiones y una muerte rápida de los mismos. En 1977 se determinó la estructura secundaria de la VFDF, amina bicíclico alcaloide, y se la renombró como anatoxina-a.[74][75] Estructuralmente es parecida a la cocaína.[76] Hay un interés continuo en anatoxina-a debido a los peligros que presenta para las aguas de recreo y el consumo, y debido a que es una molécula particularmente útil para la investigación de los receptores de acetilcolina en el sistema nervioso.[77] El carácter mortífero de la toxina significa que tiene un potencial militar elevado como un arma biológica.[78]
Curare
El término curare es ambiguo, ya que se ha utilizado para nombrar un número de venenos que en el momento de su nomenclatura fueron descritos sin los conocimientos de que disponemos hoy en día. En el pasado, las tribus de América del Sur lo utilizaban como veneno en flechas y dardos. El término ha madurado según una categorización específica de venenos que actúan en la unión neuromuscular para inhibir la señalización y, por lo tanto, inducir la relajación muscular.[79] La categoría de esta neurotoxina contiene una serie de venenos diferentes, aunque todos se produjeron originalmente de plantas de América del Sur.[79] El efecto del veneno curare inyectado se asocia generalmente a una parálisis muscular y la muerte resultante.[80] Las funciones notables del curare son la inhibición de los receptores nicotin acetilcolina en la unión neuromuscular. Normalmente, estos canales receptores permiten la entrada de iones de sodio a las células musculares para iniciar la contracción muscular. Debido al bloqueo de los receptores, la neurotoxina es capaz de reducir significativamente la señalización la unión neuromuscular, un efecto que se utilizado por los anestesistas para producir la relajación muscular.[81]
Arsénico
El arsénico es una neurotoxina que se encuentra comúnmente concentrada en áreas expuestas a la escorrentía agrícola, la minería y los sitios de fundición (Martinez-Finley 2011). Uno de los efectos de la ingestión de arsénico durante el desarrollo del sistema nervioso es la inhibición del crecimiento de neuritas[82] que puede ocurrir tanto en el sistema nervioso central como en el sistema nervioso periférico.[83] Esta inhibición del crecimiento de neuritas a menudo puede conducir a defectos de migración neuronal, y los cambios morfológicos importantes de las neuronas durante el desarrollo,[84] a menudo conduce a defectos del tubo neural en los recién nacidos. Como metabolito de arsénico, el arsenito se forma después de la ingestión de arsénico mostrando una toxicidad significativa para las neuronas a las 24 horas de exposición. El mecanismo de esta funciones de citotoxicidad a través de aumentos inducidos por el arsenito en los niveles de iones de calcio intracelular dentro de las neuronas, que pueden posteriormente reducir el potencial transmembrana mitocondrial que activa las caspasas, lo que provocó la muerte celular.[84] Otra función conocida del arsenito es su naturaleza destructiva hacia el citoesqueleto a través de la inhibición del transporte de neurofilamentos.[38] Esto es particularmente destructivo en los neurofilamentos que se utilizan en la estructura de base y apoyo de la célula. La administración de litio parece prometedora en la restauración de algunos neurofilamentos perdidos.[38] Además, de forma similar a otros tratamientos neurotóxicos, la administración de ciertos antioxidantes ha mostrado algún avance en la reducción de la neurotoxicidad del arsénico ingerido.[84]
Amoníaco
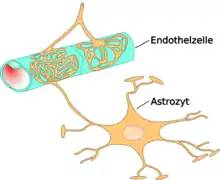
La toxicidad del amoníaco se ve a menudo a través de dos vías de administración, ya sea a través del consumo o a través de dolencias endógenas tales como la insuficiencia hepática.[85][86] Un caso notable en el que se da la toxicidad del amoníaco es la cirrosis del hígado, que se produce en la encefalopatía hepática, y puede conllevar un edema cerebral (Häussinger 2006). Este edema cerebral puede ser el resultado de la remodelación de la célula nerviosa. Como consecuencia del aumento de las concentraciones de amoníaco, la actividad “in vivo” ha demostrado que induce la inflamación de los astrocitos en el cerebro a través de aumento de la producción de cGMP (Guanosín monofosfato cíclico) dentro de las células que conducen la proteína kinasa dependiente del cGMP (PRKG) de modificación del citoesqueleto.[39] El efecto resultante de esta toxicidad es que puede reducir la energía metabólica del cerebro y su función. Es importante destacar que los efectos tóxicos del amoniaco en los astrocitos remodelados pueden reducirse a través de la administración de L-carnitina.[85] Esta remodelación de astrocitos parece estar mediada por la ammonia-induced mitochondrial permeability transition (amonio mitocondrial inducido de transición permeable). Esta transición mitocondrial es un resultado directo de la actividad de la glutamina, un compuesto que se forma a partir de amoníaco “in vivo”.[87] La administración de antioxidantes o glutaminasa inhibidor puede reducir esta transición mitocondrial y, potencialmente, también a los astrocitos remodelados.[87]
Plomo

El plomo es una potente neurotoxina cuya toxicidad ha sido reconocida durante miles de años.[88] A pesar de los efectos neurotóxicos del plomo se dan en adultos y niños de corta edad, el cerebro en desarrollo es particularmente susceptible a daño inducida por plomo, entre los efectos se pueden incluir la apoptosis y excitotoxicidad.[88] Un mecanismo subyacente por el que el plomo es capaz de causar daño es su capacidad para ser transportado por las bombas de calcio ATPasa a través de la barrera hematoencefálica, lo que permite el contacto directo con las células frágiles dentro del sistema nervioso central.[89] La neurotoxicidad de plomo se debe a la capacidad de este para actuar de una manera similar a los iones de calcio, el plomo concentrado dará lugar a una captación celular de calcio que interrumpe la homeostasis celular e induce la apoptosis.[40] Es este aumento de calcio intracelular lo que activa la proteína quinasa C (PKC), que se manifiesta como déficits de aprendizaje en niños como resultado de la exposición temprana al plomo.[40] Además de la inducción de la apoptosis, el plomo inhibe la interneurona de señalización a través de la interrupción de la liberación de neurotransmisores mediados por el calcio.[90]
Etanol
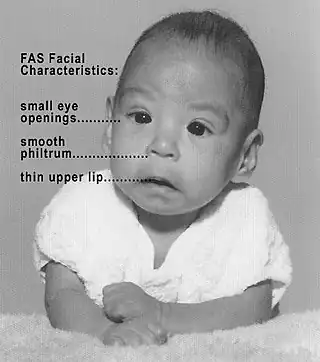
Como neurotoxina, se ha demostrado que el etanol induce daños en el sistema nervioso y puede afectar al cuerpo en una gran variedad de maneras. Los efectos conocidos a la exposición del etanol pueden ser transitorios o duraderos. Algunos de los efectos a largo plazo incluyen la neurogénesis reducida en el hipocampo,[91][92] atrofia cerebral generalizada,[93] y la inflamación inducida en el cerebro. Además, se ha demostrado que la ingestión crónica de etanol induce la reorganización de los constituyentes de la membrana celular, lo que conduce a la aparición de una bicapa lipídica marcada por el aumento de las concentraciones de colesterol y grasas saturadas.[41] Esto es importante, ya que el transporte de neurotransmisores puede verse afectado por la inhibición del transporte vesicular, dando como resultado una disminución en las funciones de la red neuronal. Un ejemplo significativo de la reducción de la comunicación entre las neuronas es la capacidad del etanol para inhibir los receptores de NMDA (N-metil D-aspartato, son receptores ionotrópicos de glutamato) en el hipocampo, lo que conlleva a una reducción de la potenciación a largo plazo, LTP (intensificación duradera en la transmisión de señales entre dos neuronas que resulta de la estimulación sincrónica de ambas) y de la adquisición de la memoria.[42] La NMDA ha demostrado desempeñar un papel importante en la LTP y, por consiguiente, en la formación de la memoria.[94] Con la ingesta crónica de etanol, aumenta la susceptibilidad de estos receptores NMDA para inducir aumentos de LTP en las neuronas mesolímbicas de dopamina en un inositol 1,4,5- trifosfato (IP3) de forma dependiente.[95] Esta reorganización puede conducir a la citotoxicidad neuronal tanto a través de la hiperactivación de las neuronas postsinápticas como por medio de la adicción inducida al consumo continuo de etanol. Se ha demostrado que el etanol reduce la acumulación de iones de calcio intracelular a través de la inhibición de la actividad del receptor de NMDA, y por lo tanto reduce la capacidad de la aparición de la LTP.[17]
Además de los efectos neurotóxicos del etanol en organismos maduros, la ingestión crónica puede producir problemas de desarrollo fetal. La primera prueba, en 1973, demostró la conexión entre el consumo crónico de etanol por las madres y los defectos en sus hijos.[96] Este trabajo fue el responsable de la clasificación de un nuevo síndrome “el Síndrome del Alcoholismo Fetal”, una enfermedad caracterizada por malformaciones comunes como defectos en la formación craneofacial, en el desarrollo de las extremidades y en las formaciones de órganos cardiovasculares. La magnitud de la neurotoxicidad de etanol en fetos que conlleva a que se dé el síndrome del alcoholismo fetal depende de cuales sean los niveles de antioxidantes en el cerebro, tales como la vitamina E.[97] Como el cerebro fetal es relativamente frágil y susceptible a tensiones inducidas, efectos nocivos graves de la exposición al alcohol se pueden apreciar en áreas importantes tales como el hipocampo y el cerebelo. La gravedad de dichos efectos dependen directamente de la cantidad y la frecuencia de consumo de etanol por la madre, y la etapa en el desarrollo del feto.[98] Se sabe que la exposición al etanol da como resultado una reducción de los niveles de antioxidantes, la disfunción mitocondrial (Chu 2007) y la muerte neuronal posterior, aparentemente como resultado de aumento en la generación de especies oxidativas reactivas (ROS).[27] Es un mecanismo plausible, ya que hay una presencia reducida en el cerebro fetal de las enzimas antioxidantes (la catalasa y la peroxidasa).[99] Para apoyar este mecanismo, la administración de altos niveles de vitamina E en preparados dietéticos reduce o incluso elimina los efectos que el etanol neurotóxico induce en los fetos.[7]
n-Hexano
El n-Hexano es una neurotoxina que ha sido responsable del envenenamiento de varios trabajadores chinos de fábricas electrónicas en los últimos años.[43][44][45][46]
Fuentes endógenas de neurotoxinas
A diferencia de la mayoría de las fuentes comunes de neurotoxinas que se adquieren por el cuerpo a través de la ingestión, las neurotoxinas endógenas pueden originarse y ejercer su función dentro del organismo vivo. Además, aunque la mayoría de los venenos y neurotoxinas exógenas rara vez poseer capacidades útiles en el organismo vivo, las neurotoxinas endógenas son comúnmente utilizadas por el cuerpo de manera útil y saludable, tales como el óxido nítrico que se utiliza en la comunicación celular. Estos compuestos endógenos son peligrosos solo cuando se encuentran en altas concentraciones.[8]
Óxido nítrico
Aunque el óxido nítrico (NO) es comúnmente utilizado por el sistema nervioso en la comunicación entre las neuronas y de señalización, puede ser activo en mecanismos que conducen a la isquemia en el cerebro (Iadecola 1998). La neurotoxicidad de NO se basa en su importancia en la excitotoxicidad del glutamato, como NO se genera en una forma dependiente de calcio en respuesta al glutamato mediada por la activación de NMDA, que se produce a una tasa elevada en excitotoxicidad del glutamato.[47] Aunque el NO facilita el aumento del flujo de sangre a regiones potencialmente isquémicas del cerebro, también es capaz de aumentar el estrés oxidativo,[100] la inducción de daño en el ADN y la apoptosis.[101] Así, un aumento de la presencia de NO en una zona isquémica del SNC puede producir efectos tóxicos de manera significativa.
Glutamato
El glutamato, como el óxido nítrico, es un compuesto producido endógenamente utilizado por las neuronas para su funcionamiento normal, está presente en pequeñas concentraciones en la materia gris del sistema nervioso central.[8] Uno de los usos más notables de glutamato endógeno es su funcionalidad como un neurotransmisor “excitatorio”.[102] Si está en altas concentraciones, el glutamato se vuelve tóxico para las neuronas circundantes. Esta toxicidad puede ser tanto un resultado de letalidad directa de glutamato en las neuronas o un resultado del flujo de calcio inducido en neuronas que conducen a inflamación y necrosis.[102] Se ha demostrado que estos mecanismos desempeñan un papel muy importante en enfermedades y complicaciones tales como la enfermedad de Huntington, la epilepsia, y el accidente cerebrovascular.[8]
Hidroxidopamina
La hidroxidopamina, o oxidopamina, es un compuesto neurotóxico orgánico sintético utilizado en investigaciones para la destrucción selectiva de neuronas dopaminérgicas y noradrenérgicas. La oxidopamina se usa mayormente en investigaciones científicas para inducir Parkinson en animales de laboratorio como ratones, ratas y monos, para desarrollar y probar nuevos medicamentos y tratamientos para la enfermedad de Parkinson. Para inducir esta condición en los animales, alrededor del 70 % de las neuronas dopaminérgicas de la sustancia negra del cerebro, deben ser destruidas, algo que se puede realizar bien con oxidopamina.
Aplicaciones en neurociencia
Pese a la diversidad de propiedades químicas y funciones, las neurotoxinas comparten la propiedad común de que actúan por algún mecanismo que conduce a la irrupción o destrucción de los componentes necesarios en el sistema nervioso. Sin embargo, las neurotoxinas, por su propio diseño, han mostrado ser muy útiles en el campo de la neurología. Dado que el sistema nervioso en la mayoría de los organismos es altamente complejo y necesario para la supervivencia, se ha convertido en un objetivo natural para el ataque tanto de depredadores como de presas. Como los organismos venenosos suelen utilizar sus neurotoxinas para someter a un depredador o una presa muy rápidamente, las toxinas han evolucionado hasta llegar a ser muy específicos para sus canales de destino de forma que la toxina no se une fácilmente a otros objetivos.[103] Como tal, las neurotoxinas proporcionan un medio eficaz por el cual ciertos elementos del sistema nervioso pueden ser elegidos como blanco de manera precisa y eficiente. Un ejemplo temprano de la elección de blanco basándose en neurotoxinas, es el uso de tetradotoxina radiomarcada para analizar los canales de sodio y obtener mediciones precisas de su concentración a lo largo de las membranas nerviosas. Del mismo modo, a través del aislamiento de ciertas actividades del canal, las neurotoxinas han proporcionado la posibilidad de mejorar el modelo de Hodgkin-Huxley de la neurona, en el cual se teorizó que únicamente los canales de sodio y potasio podían ser responsables de la mayor parte de la función del tejido nervioso.[103] A partir de este conocimiento básico, el uso de compuestos comunes tales como tetrodotoxina, tetraetilamonio y bungarotoxinas han llevado a una comprensión más profunda de las distintas maneras en que las neuronas individuales pueden comportarse.
Véase también
- Brain Facts Book at The Society for Neuroscience
- Neuroscience Texts at University of Texas Medical School
- In Vitro Neurotoxicology: An IntroductionUso incorrecto de la plantilla enlace roto (enlace roto disponible en Internet Archive; véase el historial, la primera versión y la última). at Springerlink
- Biology of the NMDA Receptor at NCBI
- Advances in the Neuroscience of Addiction, 2nd edition at NCBI
Referencias
- Sivonen, K. (1999) «Toxins produced by cyanobacteria.» Vesitalous, 5: 11-18.
- Scottish Government Blue-Green Algae (Cyanobacteria) in Inland Waters: Assessment and Control of Risks to Public Health. Consultado el 15 de diciembre de 2011.
- Spencer PS, Schaumburg HH, Ludolph AC (Eds) (2000) Experimental and Clinical Neurotoxicology. Oxford University Press, Oxford, pp. 1310.
- Olney, John W. (2002) "New Insights and New Issues in Developmental Neurotoxicology". NeuroToxicology, 23 (6): 659-68.
- Kiernan, Matthew C., Geoffrey K. Isbister, Cindy S.-Y. Lin, David Burke, and Hugh Bostock (2005) "Acute Tetrodotoxin-induced Neurotoxicity after Ingestion of Puffer Fish". Annals of Neurology, 57 (3): 339-48.
- Lidsky, Theodore I. (2003) "Lead Neurotoxicity in Children: Basic Mechanisms and Clinical Correlates". Brain. 126 (1): 5-19. Online.
- Heaton, Marieta Barrow, J. Jean Mitchell, and Michael Paiva (2000) "Amelioration of Ethanol-Induced Neurotoxicity in the Neonatal Rat Central Nervous System by Antioxidant Therapy". Alcoholism: Clinical and Experimental Research, 24 (4): 512-18.
- Choi, Dennis W (1987) "Ionic Dependence of Glutamate Neurotoxicity". The Journal of Neuroscience, 7 (2): 369-79.
- Zhang, J., V. Dawson, T. Dawson, and S. Snyder (1994) "Nitric Oxide Activation of Poly(ADP-ribose) Synthetase in Neurotoxicity". Science, 263 (5147): 687-89.
- Rosales, Raymond L., Kimiyoshi Arimura, Satoshi Takenaga, and Mitsuhiro Osame (1996) "Extrafusal and Intrafusal Muscle Effects in Experimental Botulinum Toxin-A Injection". Muscle & Nerve, 19 (4): 488-96.
- Simpson, L. L. (1986) "Molecular Pharmacology of Botulinum Toxin and Tetanus Toxin". Annual Review of Pharmacology and Toxicology, 26 (1): 427-53.
- Arnon, Stephen S., Robert Schechter, Thomas V. Inglesby, Donald A. Henderson, John G. Bartlett, Michael S. Ascher, Edward Eitzen, Anne D. Fine, Jerome Hauer, Marcelle Layton, Scott Lillibridge, Michael T. Osterholm, Tara O'Toole, Gerald Parker, Trish M. Perl, Philip K. Russell, David L. Swerdlow y Kevin Tonat (2001) "Botulinum Toxin as a Biological Weapon". The Journal of the Americal Medical Association, 285 (8): 1059-069.
- Dikranian, K (2001) "Apoptosis in the in Vivo Mammalian Forebrain". Neurobiology of Disease, 8 (3): 359-79.
- Jevtovic-Todorovic, Vesna, Richard E. Hartman, Yukitoshi Izumi, Nicholas D. Benshoff, Krikor Dikranian, Charles F. Zorumski, John W. Olney y David F. Wozniak (2003) "Early Exposure to Common Anesthetic Agents Causes Widespread Neurodegeneration in the Developing Rat Brain and Persistent Learning Deficits". The Journal of Neuroscience, 23 (3): 876-82.
- Nadler, J. Victor, Bruce W. Perry y Carl W. Cotman (1978) "Intraventricular Kainic Acid Preferentially Destroys Hippocampal Pyramidal Cells". Nature, 271 (5646): 676-77.
- Thyagarajan, B., N. Krivitskaya, J. G. Potian, K. Hognason, C. C. Garcia y J. J. McArdle (2009) "Capsaicin Protects Mouse Neuromuscular Junctions from the Neuroparalytic Effects of Botulinum Neurotoxin A". Journal of Pharmacology and Experimental Therapeutics, 331 (2): 361-71.
- Takadera, Tsuneo, Risa Suzuki y Tetsuro Mohri (1990 "Protection by Ethanol of Cortical Neurons from N-methyl-d-aspartate-induced Neurotoxicity Is Associated with Blocking Calcium Influx". Brain Research, 537(1-2): 109-14.
- Hodge, A. Trevor (2002) Roman Aqueducts and Water Supply. London: Duckworth.
- Dobbs, Michael R (2009) Clinical Neurotoxicology. Philadelphia: Saunders-Elsevier.
- Widmaier, Eric P., Hershel Raff, Kevin T. Strang, and Arthur J. Vander (2008) Vander's Human Physiology: the Mechanisms of Body Function. Boston: McGraw-Hill Higher Education.
- Martini, Frederic, Michael J. Timmons, and Robert B. Tallitsch (2009) Human Anatomy. San Francisco: Pearson/Benjamin Cummings.
- Costa, Lucio G., Gennaro Giordano, and Marina Guizzetti (2011) In Vitro Neurotoxicology: Methods and Protocols. New York: Humana.
- Harry, G. J., Melvin Billingsley, Arendd Bruinink, Iain L. Campbell, Werner Classen, David C. Dorman, Corrado Galli, David Ray, Robert A. Smith, and Hugh A. Tilson (1998) "In Vitro Techniques for the Assessment of Neurotoxicity". Environmental Health Perspectives, 106: 131–58.
- Gartlon, J., A. Kinsner, A. Balprice, S. Coecke, and R. Clothier (2006) "Evaluation of a Proposed in Vitro Test Strategy Using Neuronal and Non-neuronal Cell Systems for Detecting Neurotoxicity". Toxicology in Vitro, 20 (8): 1569-581.
- Mundy 2008
- Lotti, Marcello, and Angelo Moretto (1989) "Organophosphate-Induced Delayed Polyneuropathy". Toxicological Reviews, 24 (1) (2005): 37–49.
- Brocardo, Patricia S., Joana Gil-Mohapel, and Brian R. Christie (2011) "The Role of Oxidative Stress in Fetal Alcohol Spectrum Disorders". Brain Research Reviews, 67 (1–2): 209–25.
- Lewendon, G., S. Kinra, R. Nelder, and T. Cronin (2001) "Should Children with Developmental and Behavioural Problems Be Routinely Screened for Lead?". Archives of Disease in Childhood, 85: 286–88.
- Haghdoost-Yazdi, Hashem, Ayda Faraji, Negin Fraidouni, Mohadeseh Movahedi, Elham Hadibeygi, and Fatemeh Vaezi (2011) "Significant Effects of 4-aminopyridine and Tetraethylammonium in the Treatment of 6-hydroxydopamine-induced Parkinson’s Disease". Behavioural Brain Research, 223: 70–74.
- Debin, John A., John E. Maggio, and Gary R. Strichartz (1993) "Purification and Characterization of Chlorotoxin, a Chloride Channel Ligand from the Venom of the Scorpion". The American Physiological Society, pp. 361–69.
- McCleskey, E. W. (1987) "Omega-conotoxin: Direct and Persistent Blockade of Specific Types of Calcium Channels in Neurons but Not Muscle". Proceedings of the National Academy of Sciences, 84 (12): 4327–331.
- Garcia-Rodriguez, C., I. N. Geren, J. Lou, F. Conrad, C. Forsyth, W. Wen, S. Chakraborti, H. Zao, G. Manzanarez, T. J. Smith, J. Brown, W. H. Tepp, N. Liu, S. Wijesuriya, M. T. Tomic, E. A. Johnson, L. A. Smith, and J. D. Marks (2011) "Response Re: 'Neutralizing Human Monoclonal Antibodies Binding Multiple Serotypes of Botulinum Neurotoxin' by Garcia-Rodriguez Et Al., PEDS, 2011;24:321–331". Protein Engineering Design and Selection, 24 (9): 633–34.
- Williamson, Lura C., Jane L. Halpern, Cesare Montecucco, J. E. Brown, and Elaine A. Neale (1996) "Clostridial Neurotoxins and Substrate Proteolysis in Intact Neurons". The Journal of Biological Chemistry, 271 (13): 7694–699.
- Dutrere 2006
- Koller 1988
- Banks, William A., and Abba J. Kastin (1989) "Aluminum-Induced Neurotoxicity: Alterations in Membrane Function at the Blood-Brain Barrier". Neuroscience & Biobehavioral Reviews, 13: 47–53.
- Aschner, M., and J. Aschner (1990) "Mercury Neurotoxicity: Mechanisms of Blood-brain Barrier Transport". Neuroscience & Biobehavioral Reviews, 14 (2): 169–76.
- DeFuria, Jason (2006) "The Environmental Neurotoxin Arsenic Impairs Neurofilament Dynamics by Overactivation of C-JUN Terminal Kinase: Potential Role for Amyotrophic Lateral Sclerosis". UMI, pp. 1–16.
- Konopacka, Agnieszka, Filip A. Konopacki, and Jan Albrecht (2009) "Protein Kinase G Is Involved in Ammonia-induced Swelling of Astrocytes". Journal of Neurochemistry, 109: 246–51.
- Bressler J, Kim KA, Chakraborti T, Goldstein G (1999) Molecular mechanisms of lead neurotoxicity. [Review]. Neurochem Res, 24: 595-600.
- Leonard, B. E. (1986) "Is Ethanol a Neurotoxin?: the Effects of Ethanol on Neuronal Structure and Function". Alcohol and Alcoholism, 21 (4): 325-38.
- Lovinger, D., G. White y F. Weight. "Ethanol Inhibits NMDA-activated Ion Current in Hippocampal Neurons". Science, 243 (4899): 1721-724.
- Workers poisoned while making iPhones ABC News Octover 25 2010
- Dirty Secrets ABC Foreign Correspondent 2010-Oct-26
- Mr Daisey and the Apple Factory, This American Life January 6 2012
- Occupational Safety and Healt Guideline for n-Hexane OSHA.gov
- Garthwaite, John, Sarah L. Charles, and Russel Chess-Williams (1988) "Endothelim-derived Relaxing Factor Release on Activation of NMDA Receptors Suggests Role as Intercellular Messenger in the Brain". Nature, 336 (24): 385–88.
- Chowdhury, F. R., H A M. Nazmul Ahasan, A K M. Mamunur Rashid, A. Al Mamun, and S. M. Khaliduzzaman (2007) "Tetrodotoxin Poisoning: a Clinical Analysis, Role of Neostigmine and Short-term Outcome of 53 Cases". Singapore Medical Journal, 48 (9): 830–33.
- Ahasan, H A M N, A. A. Mamun, S. R. Karim, M. A. Baker, E. A. Gazi, and C. S. Bala (2004) "Paralytic Complications of Puffer Fish (Tetrodotoxin) Poisoning". Singapore Medical Journal, 73 (42.2): 73–74.
- Lau, F. L., C. K. Wong, and S. H. Yip (1995) "Puffer Fish Poisoning". Emergency Medicine Journal, 12 (3): 214–15.
- Stanfield, Peter R (1983) "Tetraethylammonium Ions and the Potassium Permeability of Excitable Cells". Reviews of Physiology, Biochemistry & Pharmacology, 97: 1–49.
- Roed, A. (1989) "The Effects of Tetraethylammonium during Twitch and Tetanic Stimulation of the Phrenic Nerve Diaphragm Preparation in the Rat". Neuropharmacology, 28 (6): 585–92.
- Deshane, Jessy, Craig C. Garner, and Harald Sontheimer (2003) "Chlorotoxin Inhibits Glioma Cell Invasion via Matrix Metalloproteinase-2". The Journal of Biological Chemistry, 278 (6): 4135–144.
- Soroceanu, Liliana, Yancey Gillespie, M. B. Khazaeli, and Harold Sontheimer (1998) "Use of Chlorotoxin for Targeting of Primary Brain Tumors". Cancer Research, 58: 4871–879
- Jacob, Reed B., and Owen M. McDougal (2010) "The M-superfamily of Conotoxins: a Review". Cellular and Molecular Life Sciences, 67: 17–27.
- Olivera, Baldomero M., Lourdes J. Cruz, Victoria De Santos, Garth LeCheminant, David Griffin, Regina Zeikus, J. Michael McIntosh, Robert Galyean, and Janos Varga (1987) "Neuronal Calcium Channel Antagonists. Discrimination between Calcium Channel Subtypes Using.omega.-conotoxin from Conus Magus Venom". Biochemistry, 26 (8): 2086–090.
- Cruz, Lourdes J., and Baldomero M. Olivera (1987) "Calcium Channel Antagonists ω-Conotoxin Defines a New High Affinity Site". The Journal of Biological Chemistry, 14 (261): 6230–233.
- Brin, Mitchell F (1997) "Botulinum Toxin: Chemistry, Pharmacology, Toxicity, and Immunology". Muscle & Nerve, 20 (S6): 146–68.
- Montecucco, C. (1986) "How Do Tetanus and Botulinum Toxins Bind to Neuronal Membranes?"Trends in Biochemical Sciences 11.8: 314–17.
- Pirazzini, Marco, Ornella Rossetto, Paolo Bolognese, Clifford C. Shone, and Cesare Montecucco (2011) "Double Anchorage to the Membrane and Intact Inter-chain Disulfide Bond Are Required for the Low PH Induced Entry of Tetanus and Botulinum Neurotoxins into Neurons". Cellular Microbiology: No. Print.
- King, Steven W., John Savory, Michael R. Wills, and H. J. Gitelman (1981) "The Clinical Biochemistry of Aluminum". Critical Reviews in Clinical Laboratory Sciences, 14 (1): 1–20.
- Rabe, Ausma, Moon He Lee, Judy Shek, and Henryk M. Wisniewski (1982) "Learning Deficit in Immature Rabbits with Aluminum-induced Neurofibrillary Changes". Experimental Neurology, 76 (2): 441–46.
- Walton, J. (2006) "Aluminum in Hippocampal Neurons from Humans with Alzheimer's Disease". NeuroToxicology, 27 (3): 385–94.
- Chan, H. M. (2011) "Mercury in Fish: Human Health Risks". Encyclopedia of Environmental Health: 697–704.
- Brookes, N (1988) "Specificity and Reversibility of the Inhibition by HgCl of Glutamate Transport in Astrocyte Cultures". Journal of Neurochemistry, 50 (4): 1117–122.
- Tsetlin, V.I, and F. Hucho (2004) "Snake and Snail Toxins Acting on Nicotinic Acetylcholine Receptors: Fundamental Aspects and Medical Applications". FEBS Letters, 557 (1–3): 9–13.
- Liu, Kuang-Kai, Mei-Fang Chen, Po-Yi Chen, Tony J F. Lee, Chia-Liang Cheng, Chia-Ching Chang, Yen-Peng Ho, and Jui-I Chao (2010) "Alpha-bungarotoxin Binding to Target Cell in a Developing Visual System by Carboxylated Nanodiamond". Nanotechnology, 19 (20): 205102.
- Hue, Bernard, Steven D. Buckingham, David Buckingham, and David B. Sattelle (2007) "Actions of Snake Neurotoxins on an Insect Nicotinic Cholinergic Synapse". Invertebrate Neuroscience, 7 (3): 173–78.
- Carmichael WW, Biggs DF, Gorham PR (1975). "Toxicology and pharmacological action of Anabaena flos-aquae toxin". Science 187 (4176): 542–544. doi:10.1126/science.803708. PMID 803708.
- Carmichael WW, Gorham PR (1978). "Anatoxins from clones of Anabaena flos-aquae isolated from lakes of western Canada". Mitt. Infernal. Verein. Limnol"., 21: 285–295.
- Yang, X (2007) Occurrence of the cyanobacterial neurotoxin, anatoxin-a, in New York State waters ProQuest. ISBN 978-0-549-35451-2.
- Wood S. A., Rasmussen J. P., Holland P. T., Campbell R., Crowe A. L. M. (2007). "First Report of the Cyanotoxin Anatoxin-A from Aphanizomenon issatschenkoi (cyanobacteria)". Journal of Phycology 43 (2): 356–365. doi:10.1111/j.1529-8817.2007.00318.x.
- National Center for Environmental Assessment (2006) "Toxicological Reviews of Cyanobacterial Toxins: Anatoxin-a" NCEA-C-1743
- Devlin JP, Edwards OE, Gorham PR, Hunter NR, Pike RK, Stavric B (1977). "Anatoxin-a, a toxic alkaloid from Anabaena flos-aquae NRC-44h". Can. J. Chem. 55 (8): 1367–1371. doi:10.1139/v77-189.
- Moore RE (1977). "Toxins from blue-green algae". BioScience 27 (12): 797–802. doi:10.2307/1297756. JSTOR 1297756.
- Metcalf JS and Codd GA (2009) "Cyanobacteria, neurotoxins and water resources: Are there implications for human neurodegenerative disease?" Informa Healthcare, 10(s2): 74–78.
- Stewart I, Seawright AA, Shaw GR (2008). "Cyanobacterial poisoning in livestock, wild mammals and birds – an overview" (PDF). Cyanobacterial Harmful Algal Blooms: State of the Science and Research Needs. Advances in Experimental Medicine and Biology 619: 613–637. doi:10.1007/978-0-387-75865-7_28. ISBN 978-0-387-75864-0.
- Dixit A, Dhaked RK, Alam SI, Singh L (2005). "Military potential of biological neurotoxins". Informa Healthcare 24 (2): 175–207. doi:10.1081/TXR-200057850.
- Bisset, Norman G (1992) "War and Hunting Poisons of the New World. Part 1. Notes on the Early History of Curare". Journal of Ethnopharmacology, 36 (1): 1–26.
- Schlesinger, Edward B. (1946) "Curare A Review of Its Therapeutic Effects and Their Physiological Basis". The American Journal of Medicine, 1 (5): 518–30.
- Griffith, Harold R., and G. Enid Johnson (1942) "The Use Of Curare In General Anesthesia". Anesthesiology, 3 (4): 418–420.
- Liu, Kuang-Kai, Mei-Fang Chen, Po-Yi Chen, Tony J F. Lee, Chia-Liang Cheng, Chia-Ching Chang, Yen-Peng Ho, and Jui-I Chao (2010) "Alpha-bungarotoxin Binding to Target Cell in a Developing Visual System by Carboxylated Nanodiamond". Nanotechnology, 19 (20): 205102.
- Vahidnia, A., G.B. Van Der Voet, and F.A. De Wolff (2007) "Arsenic Neurotoxicity A Review". Human & Experimental Toxicology, 26 (10): 823–32.
- Rocha, R. A., J. V. Gimeno-Alcaniz, Raymond Martín–Ibanez, J. M. Canals, D. Vélez, and V. Devesa (2011) "Arsenic and Fluoride Induce Neural Progenitor Cell Apoptosis". Toxicology Letters, 203: 237–44.
- Matsuoka, Masato, Hideki Igisu, Kazuaki Kohriyama y Naohide Inoue (1991) "Suppression of Neurotoxicity of Ammonia by L-carnitine". Brain Research, 567 (2): 328-31.
- Buzanska, L., B. Zablocka, A. Dybel, K. Domanska-Janik y J. Albrecht (2000) "Delayed Induction of Apoptosis by Ammonia in C6 Glioma Cells". Neurochemistry International, 37: 287–97.
- Norenberg, M. D., K. V. Rama Rao y A. R. Jayakumar (2004) "Ammonia Neurotoxicity and the Mitochondrial Permeability Transition". Journal of Bioenergetics and Biomembranes, 36 (4): 303-07.
- Lidsky, Theodore I. (2003) "Lead Neurotoxicity in Children: Basic Mechanisms and Clinical Correlates". Brain. 126 (1): 5-19. Online.
- Bradbury MW, Deane R. Permeability of the blood±brain barrier to lead. [Review]. Neurotoxicology, 14: 131–6.
- Lasley SM, Green MC, Gilbert ME (1999) "Influence of exposure period on in vivo hippocampal glutamate and GABA release in rats chronically exposed to lead". Neurotoxicology, 20: 619–29.
- Taffe, M. A., R. W. Kotzebue, R. D. Crean, E. F. Crawford, S. Edwards, and C. D. Mandyam (2010) "From the Cover: Long-lasting Reduction in Hippocampal Neurogenesis by Alcohol Consumption in Adolescent Nonhuman Primates". Proceedings of the National Academy of Sciences, 107 (24): 11104–1109.
- Morris, Stephanie A., David W. Eaves, Aleksander R. Smith y Kimberly Nixon (2009) "Alcohol Inhibition of Neurogenesis: A Mechanism of Hippocampal Neurodegeneration in an Adolescent Alcohol Abuse Model". Hippocampus: NA.
- Bleich, S (2003) "Hyperhomocysteinemia as a New Risk Factor for Brain Shrinkage in Patients with Alcoholism". Neuroscience Letters, 335 (3): 179–82.
- Davis, S., S. P. Butcher, and R. Morris (1992) "The NMDA Receptor Antagonist D-2-amino-5phosphonopentanoate (D-AP5) Impairs Spatial Learning and LTP in Vivo at Lntracerebral Concentrations Comparable to Those That Block LTP in Vitro". The Journal of Neuroscience, 12 (1): 21–34.
- Bernier, Brian E., Leslie R. Whitaker y Hitoshi Morikawa (2011) "Previous Ethanol Experience Enhances Synaptic Plasticity of NMDA Receptors in the Ventral Tegmental Area". The Journal of Neuroscience, 31.14: 5305–212.
- Jones, K. (1973) "Pattern Of Malformation In Offspring Of Chronic Alcoholic Mothers". The Lancet, 301 (7815): 1267–271.
- Mitchell, J. Jean, Michael Paiva y Marieta Barrow Heaton (1999) "The Antioxidants Vitamin E and β-Carotene Protect Against Ethanol-Induced Neurotoxicity in Embryonic Rat Hippocampal Cultures". Alcohol, 17 (2): 163-68.
- Gil-Mohapel, Joana, Fanny Boehme, Leah Kainer y Brian R. Christie (2010) "Hippocampal Cell Loss and Neurogenesis after Fetal Alcohol Exposure: Insights from Different Rodent Models". Brain Research Reviews, 64 (2): 283-303.
- Bergamini, Carlo M., Stefani Gambetti, Alessia Dondi y Carlo Cervellati (2004) "Oxygen, Reactive Oxygen Species and Tissue Damage". Current Pharmaceutical Design, 10 (14): 1611-626.
- Beckman, J. S. (1990) "Apparent Hydroxyl Radical Production by Peroxynitrite: Implications for Endothelial Injury from Nitric Oxide and Superoxide". Proceedings of the National Academy of Sciences, 87 (4): 1620-624.
- Bonfoco, E (1993) "Apoptosis and Necrosis: Two Distinct Events Induced, Respectively, by Mild and Intense Insults with N-Methyl-D-Aspartate or Nitric Oxide/Superoxide in Cortical Cell Cultures". Proceedings of the National Academy of Sciences 92.16 (1995): 7162-166.
- Choi, D. W. y S. M. Rothman (1990) "The Role of Glutamate Neurotoxicity in Hypoxic-Ischemic Neuronal Death". Annual Review of Neuroscience, 13 (1): 171-82.
- Adams, Michael E., and Baldomero M. Olivera (1994) "Neurotoxins: Overview of an Emerging Research Technology". Trends in Neuroscience, 17 (4): 151-55.
Enlaces externos
 Wikimedia Commons alberga una categoría multimedia sobre Neurotoxina.
Wikimedia Commons alberga una categoría multimedia sobre Neurotoxina.- Environmental Protection Agency at United States Environmental Protection Agency
- Alcohol and Alcoholism at Oxford Medical Journals
- Neurotoxicology at Elsevier Journals
- Neurotoxin Institute at Neurotoxin Institute
- Neurotoxins Archivado el 23 de octubre de 2013 en Wayback Machine. at Toxipedia
| Control de autoridades |
|
|---|
 Datos: Q407752
Datos: Q407752- Multimedia: Neurotoxins / Q407752