PCR en tiempo real
La PCR cuantitativa (en inglés, quantitative polymerase chain reaction; qPCR o Q-PCR) o PCR en tiempo real (en inglés real time PCR) es una variante de la reacción en cadena de la polimerasa (PCR) utilizada para amplificar y simultáneamente cuantificar de forma absoluta el producto de la amplificación de ácido desoxirribonucleico (ADN). Para ello emplea, al igual que la PCR convencional, un molde de ADN, al menos un par de cebadores específicos, dNTPs, un tampón de reacción adecuado y una ADN polimerasa termoestable. A dicha mezcla se le adiciona una sustancia marcada con un fluoróforo que permita medir la tasa de generación de uno o más productos específicos.[1] en un termociclador provisto de sensores de fluorescencia, tras excitar el fluoróforo a la longitud de onda apropiada. Dicha medición se realiza tras cada ciclo de amplificación, y es por esto que se le denomina PCR en tiempo real (es decir, PCR inmediata, simultánea). En muchos casos, el molde que se emplea para la PCR cuantitativa no es ADN desde el principio, sino que puede ser ADN complementario (ADNc), de hebra simple, obtenido por retrotranscripción de ácido ribonucleico (ARN); en este caso, la técnica es una RT-PCR cuantitativa o en tiempo real, o RT-Q-PCR. Debe evitarse la confusión con la técnica denominada «PCR tras transcripción inversa» (RT-PCR, del inglés reverse transcriptase PCR), en la cual existe un paso de retrotranscripción de ARN a ADN pero que no necesariamente cuantifica el producto a tiempo real.
La PCR cuantitativa junto a los chip de ADN son modernas metodologías para el estudio de la expresión génica, aunque otros métodos tradicionales como el Northern blot, si bien no con tanta precisión, también permiten su abordaje.[2] La variabilidad que se introduce en la cuantificación y que conlleva a errores en la estimación deriva de la integridad del ADN, la eficiencia enzimática y otros muchos factores, por lo que se han desarrollado numerosos sistemas de estandarización. Los hay para cuantificar de forma absoluta la expresión génica, pero, de forma más común, se orientan a la cuantificación relativa del gen de estudio respecto de otro, denominado «normalizador», que se selecciona por su expresión casi constante. Estos genes suelen denominarse, en inglés, house-keeping genes, ya que suelen estar involucrados en funciones básicas en la supervivencia celular, lo cual suele implicar una expresión constitutiva.[3][4] De este modo, efectuando en cada experimento la medición de los genes de interés y dividiéndolos por la expresión del gen normalizador seleccionado es posible comparar los primeros aún sin conocer en términos absolutos su nivel de expresión. Los genes normalizadores más empleados son aquellos que codifican las siguientes proteínas: tubulina, gliceraldehído-3-fosfato deshidrogenasa, albúmina, ciclofilina, ARN ribosomales, etc.[2]
Fundamento
La PCR cuantitativa se realiza en un termociclador con capacidad de hacer incidir sobre cada muestra un haz de luz de una longitud de onda determinada y de detectar la fluorescencia emitida por el fluorocromo excitado. Este termociclador es un aparato con capacidad para calentar y enfriar rápidamente las muestras, de modo que se aprovechen las cualidades fisicoquímicas de los ácidos nucleicos y las enzimáticas de la ADN polimerasa.
El proceso de PCR, por lo general, consiste en una serie de cambios de temperatura que se repiten 25-40 veces, llamados ciclos, cada uno con un mínimo de tres etapas: la primero, en torno a los 95 °C, permite la separación de los ácidos nucleicos de doble cadena; la segunda, a una temperatura en torno a los 50-60 °C, permite el alineamiento de los cebadores al ADN molde;[5] la tercera, a 68-72 °C, facilita la polimerización por parte de la ADN polimerasa. Dado el pequeño tamaño de los fragmentos amplificados usualmente en este tipo de PCR puede omitirse el último paso, pues la enzima es capaz de amplificar durante la rampa entre la temperatura de alineamiento y la de desnaturalización. Además, algunos termocicladores añaden a cada ciclo unos segundos a otra temperatura, por ejemplo 80 °C, a fin de reducir el ruido por la presencia de dímeros de cebadores cuando se emplea un colorante inespecífico. Las temperaturas usadas y el tiempo aplicado en cada ciclo dependen de gran variedad de parámetros, tales como la enzima usada para la síntesis de ADN, la concentración de iones divalentes y desoxirribonucleótidos (dNTPs) en la reacción o la temperatura de unión de los cebadores.[6]
Clasificación
Podemos clasificar las técnicas de PCR cuantitativa según el empleo de fluorocromos no específicos o bien de sondas moleculares dependientes de la secuencia.
- En las técnicas basadas en fluorocromos inespecíficos se detecta la generación exponencial de ADN de doble cadena empleando un fluorocromo que se une inespecíficamente a aquel. Un ejemplo de colorante que permite esta detección es el SYBR Green (Verde SYBR) que, excitado mediante luz azul (λmax = 488 nm) emite luz verde (λmax = 522 nm).[7] Posee la ventaja de requerir solo un par de cebadores para efectuar la amplificación, lo que abarata su coste; sin embargo, solo es posible amplificar un producto en cada reacción.
- Las técnicas basadas en sondas específicas utilizan al menos un oligonucleótido marcado fluorescentemente. Típicamente esta sonda está unida a dos fluorocromos e hibrida en la zona intermedia entre el cebador directo (forward) y el inverso (reverse); esto es, en el amplicón. De este modo, cuando la sonda está intacta, se produce transferencia de energía por resonancia (FRET). Dicha FRET no se produce cuando los dos fluorocromos están distantes debido a la degradación de la sonda mediante la actividad 5'-3' exonucleasa de la ADN polimerasa, o bien debido a la separación física de los fluorocromos por un cambio en la conformación de la sonda. Esto permite monitorizar el cambio del patrón de fluorescencia y deducir el nivel de amplificación del gen.
Análisis de temperatura de fusión
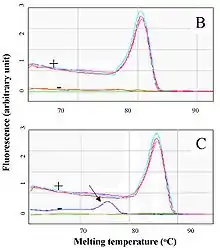
La Q-PCR permite, empleando un fluorocromo de unión inespecífica a la doble hebra de ADN (generalmente SYBR Green) identificar fragmentos amplificados de ADN concretos a partir de la temperatura de fusión (también denominado valor Tm, del inglés melting temperature), específica para el fragmento amplificado que se está buscando, y cuyos resultados se obtienen a partir de la observación de la curva de disociación de las muestras de ADN analizadas.[9]
Ello permite, a diferencia de PCR convencional, prescindir del posterior empleo de técnicas de electroforesis para la visualización de los resultados de todas las muestras. Porque, pese a que la PCR cuantitativa es una técnica cinética, suele ser evaluada a punto final. Así, esta técnica lleva a la obtención de resultados más rápidos o con menor gasto de reactivos empleados en las técnicas de electroforesis. No obstante, según el criterio del investigador, posteriormente puede ser necesario correr en geles las muestras cuyo resultados previos en el PCR en tiempo real se pueden considerar dudosos o para ratificar resultados en muestras positivas.
Cuantificación de la expresión génica
La cuantificación puede realizarse en términos absolutos o relativos. En el primer caso, la estrategia es relacionar la señal de amplificación obtenida con el contenido en ADN empleando una curva de calibrado; para este enfoque es vital que la PCR de la muestra y de los elementos de la recta de calibrado posean una misma eficiencia de amplificación. En el segundo caso, se expresa el cambio en los niveles de expresión de ARN mensajero (ARNm) interpretado como ADN complementario (ADNc, generado por retrotranscripción del ARNm); esta cuantificación relativa es más fácil de realizar, puesto que no requiere curva de calibrado, y se sustenta en la comparación entre el nivel de expresión del gen a estudiar versus un gen control (también llamado de referencia, interno o normalizador o, en inglés, housekeeping gene).
Por tanto, en la cuantificación relativa es irrelevante en qué unidades se expresa la cuantificación, y sus resultados son comparables entre múltiples experimentos de RT-Q-PCR. De hecho, el propósito de emplear uno o más genes de normalización es corregir la variación no específica, como las diferencias en la cantidad y calidad del ARN empleado, que pueden afectar a las eficiencias de la retrotranscripción y PCR. No obstante, el aspecto crucial es que la estabilidad del gen de referencia sea una realidad.[10]
La selección de los genes internos se ha realizado clásicamente en Biología Molecular analizando la estabilidad de la expresión en estudios cualitativos o de baja sensibilidad, como el examen visual de geles de ARN, densitometría de Northern blots o PCR semicuantitativa (PCR mimic). En plena era de la genómica, es posible realizar una aproximación a gran escala empleando los chips de ADN para muchos organismos.[11] No obstante, se ha descrito que la mayoría de los genes empleados como normalizadores en la cuantificación de la expresión de ARN mensajero varían según las condiciones experimentales.[12][13][14] Por ello, es preciso realizar un estudio metodológico previo a fin de seleccionar, con ayuda de herramientas estadísticas, los más apropiados.
Se han desarrollado varios algoritmos estadísticos que detectan qué gen o genes son los más apropiados para la normalización de un conjunto de tejidos bajo unas condiciones dadas: algunos, como geNORM o BestKeeper, realizan comparaciones por pares y medias geométricas sobre una matriz de expresión de genes de referencia para diversos tejidos.[15][16]
Modelización
A diferencia de la PCR de punto final (PCR convencional), la PCR en tiempo real permite cuantificar el nivel de producto obtenido en cualquier momento de la amplificación mediante la señal de fluorescencia (en realidad, mediante su nivel sobre un umbral). Los valores de fluorescencia, expresados como logaritmos para estudiar fácilmente la fase exponencial de amplificación, aparecen como una línea recta al representarlos gráficamente frente al número de ciclo; este segmento, denominado segmento cuantificable, permite valorar la cantidad de ADN inicial.
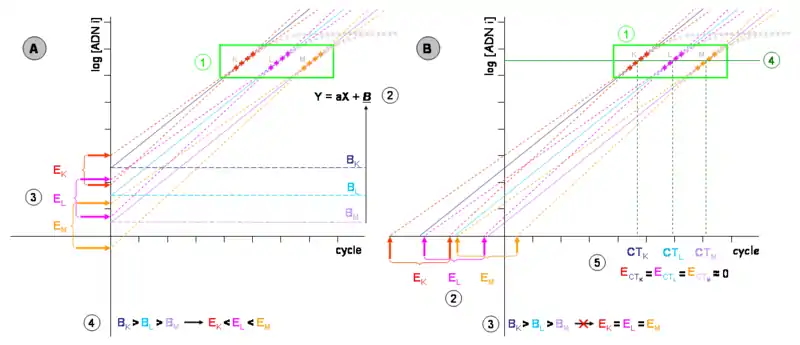
- Gráfica de la izquierda (A)
Las cinéticas de tres PCR de tres muestras distintas (K, L y M) de concentraciones de ADN inicial decrecientes (concretamente, se reducen a la décima parte cada vez) se representan en un gráfico semilogarítimo de fluorescencia (en ordenadas) frente al ciclo de PCR (en abscisas). No se representan las fases previas a la exponencial.
- Zona de segmentos cuantificables de cada muestra. En gris la zona de saturación, fuera ya de la zona lineal.
- Cada uno de los segmentos cuantificables permite definir una ecuación de la recta de tipo y=ax+b, que permite modelizar la eficiencia de la amplificación (la pendiente a) y, mediante la ordenada en el origen b, la cantidad de ADN en el ciclo 0, al menos teóricamente.
- Dicha cuantificación arrastra un error estocástico, asociado a toda medidas experimental. Si las muestras de concentraciones K, L y M fueran amplificadas durante más ciclos, se obtendrían cinéticas diferentes, aunque bastante próximas (se representan sus segmentos cuantificables en rojo, rosa y naranja). Cada una de estas medidas permite establecer una nueva ecuación, representada con puntos del mismo color. Las ordenadas en el origen (b) diferentes son también medidas. Así, podemos evaluar el error o incertidumbre de la técnica evaluando las diferencias entre las diferentes b (EK, EL, EM).
- No obstante, dicha imprecisión en la medida varía para cada muestra (EK es inferior a EL, que es, a su vez, más pequeña que EM). Una proyección de la ecuación de la derecha sobre el eje de ordenadas o una de sus paralelas dará lugar a un «error dependiente de la concentración de ADN inicial».
- Gráfica de la derecha (B)
- Las ecuaciones obtenidas según los segmentos cuantificables pueden extrapolarse hacia el eje de abscisas, aunque se trate de un valor carente de significado bioquímico sino simplemente matemático.
- Los ángulos que modelizan el error experimental (líneas de puntos rojos, naranjas o rosas) permiten definir las nuevas incertidumbres EK, EL y EM. Estas incertidumbres son mayores que en la gráfica de la izquierda (gráfica A), si bien se mantiene su proporcionalidad. Por tanto, sobre la medida ahora generamos un «error independiente de la concentración de ADN inicial».
- Es posible proyectar rectas paralelas entre sí, sobre la recta definida sobre una paralela al eje de abscisas de modo que se corten los segmentos cuantificables por la mitad. Este segmento es denominado «umbral de detección».
- Los valores en X (número de ciclos) de estas intersecciones son denominados «CT» (o «Ct», del inglés cycle threshold, cuya traducción es ciclo umbral), aunque también pueden llamarse «CP» (del inglés crossing point, o punto de cruce). Se trata, pues, de los valores matemáticos definidos sobre el espacio de reales positivos y no de los enteros positivos (aunque una fracción del ciclo no posea realidad experimental). Estos valores, inversamente proporcionales a la cantidad de ADN inicial, suelen poseer una incertidumbre sobre la medida mínima, en general inferior al 5 %.
Rectas de calibrado
Como se indicaba en el apartado de cuantificación, el empleo del CT como valor matemático permite obtener resultados fiables, pero este hecho puede no ser explotado directamente. A fin de conocer la cantidad de ADN inicial, es preciso pues realizar nuevas transformaciones matemáticas que requieren conocer la eficiencia de PCR, que suele determinarse gracias a una recta de calibrado.
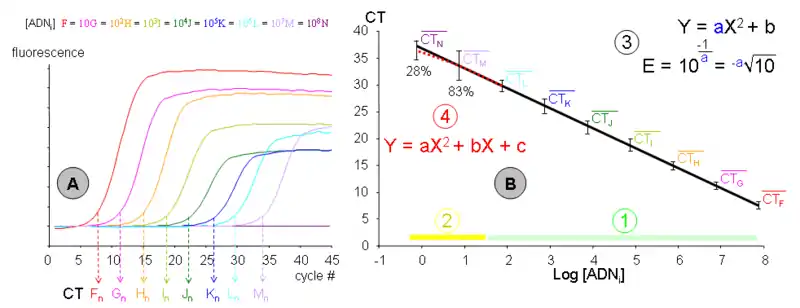
- Gráfica de la izquierda (A)
Las nuevas muestras F, G, H, I, J, K, L, M y N, de concentración de ADN inicial decreciente (un orden de magnitud cada vez) son amplificadas mediante PCR en un mismo experimento. Cada cinética permite determinar un CT para cada uno (es decir, un número en referencia a un ciclo en concreto). Se representan las concentraciones de ADN como número de moléculas por tubo (en este caso, F posee 70 millones y N, 0,7).
Los rendimientos representados corresponden a «n» reacciones, cuya fiabilidad se debe a la existencia de réplicas independientes en la reacción, personal y reactivos. La fluorescencia se muestra en unidades arbitrarias y el ruido de fondo ha sido limpiado. Cabe destacar que la muestra Nn no ha amplificado en absoluto.
Los valores CT medios en función de la cantidad de ADN inicial de todas las réplicas pueden representarse en un gráfico semilogarítmico. De este modo, la recta de calibrado para estos valores medios puede definirse gracias a una regresión lineal con un determinado coeficiente de correlación (r2) que, para ser considerado de calidad, debe poseer un valor mínimo de 0,9999. No obstante, han de representarse los errores, generalmente definidos según el rango de valores obtenidos para un punto.
Con estos datos, es posible estudiar:
- La fase «cuantitativa y detectable de la PCR». En la cual todas las muestras son detectables y se alinean en todos sus puntos con su recta particular. Generalmente, esta fase comprende las concentraciones de ADN iniciales en torno a 102 o 108 copias del ácido nucleico. Por debajo de estas cifras los fenómenos estocásticos alteran perceptiblemente los resultados, si bien es posible compensarlos con un alto número de réplicas en las medidas. Por encima de estos valores, el «ruido de fondo» es suficientemente bajo como para no poder ser determinado; en este caso, es posible compensar esta deficiencia mediante un protocolo de PCR con una eficiencia muy alta o, de forma más simple, diluyendo la muestra inicial.
- La fase «a veces detectable pero no cuantitativa de la PCR». Que comprende las concentraciones de ADN inicial entre una copia y la decena de copias. El porcentaje de muestras medidas, en el ejemplo de la gráfica, se indica mediante las concentraciones M y N, o sea 83 % para una concentración media de siete copias y 28 % cuando tres de los cuatro tubos contienen una copia (0,7 de concentración, o –0,15 en logaritmo). La dispersión de las medidas, y por ello el margen de error, se incrementan perceptiblemente. Nótese que un buen número de medidas M y N no se alinean con su recta particular, aunque no lo hagan por motivos de azar. Por tanto, solo replicando más veces las medidas es posible determinar con precisión esta fase.
- La recta de calibrado, por tanto, representada en un gráfico semilogarítmico da lugar a una recta definida por la fórmula Y = aX + B donde:
- Y es el CT medido por el termociclador.
- La pendiente a deriva de la eficiencia de la PCR, y puede ser calculada mediante la ecuación
- Nótese que en el esquema se representa el caso más frecuente en el que la concentración está expresada en logaritmos decimales. Esta pendiente a menudo se considera una constante de amplificación para cada gen, par de cebadores y condición de PCR particular. Por ello esta pendiente o eficiencia de amplificación se emplea para cuantificar en la PCR cuantitativa.
- X es la concentración de ADN inicial expresada en logaritmo de copias/tubo, ng/µL, unidades arbitrarias, etc.
- B es un punto matemático sin realidad experimental (log 0 = 1/∞) que, sin embargo, puede ser empleado para calibrar cada experimento de PCR (los «runs», en inglés) con sus semejantes. De este modo, si las rectas de calibrado lo fueron por su intersección al origen B, la dispersión será menor, salvo para los puntos M y N.
![{\displaystyle E=(indice\;del\;logaritmo)^{\frac {-1}{pendiente}}={\sqrt[{-pendiente}]{indice\;del\;logaritmo}}}](../I/a62e8b7798886375544d6e9f3b6238fc2567ad75.svg)
- La recta de regresión no pasa por el punto central de la dispersión en cada medida para las concentraciones M y N, pero sí según la pendiente y un factor de «amortiguación». Esto es: si consideramos CT medios para L y N serán modelizados de mejor manera mediante un polinomio de segundo grado. El software de algunos termocicladores permite tener en cuenta esta «amortiguación», si bien hay que tener en cuenta que:
- Dicha amortiguación es extremadamente variable de una recta de calibrado a otra, y la corrección que se modeliza no corresponde probablemente a aquella que pasa por la muestra cuantificada. El software comercial lo modeliza para una sola muestra.
- La imprecisión en la cuantificación a estas concentraciones es tan importante que la modelización de la «amortiguación» puede ser necesaria.
- Esta «amortiguación» corresponde, cuando se dibujan las rectas, a una reducción de la pendiente luego de un aumento de la eficacia de PCR, así que el incremento de los efectos estocásticos puede reducirse. Nótese además que esta «amortiguación» se define principalmente según la concentración más baja (N, 0,7 copias en el ejemplo). O lo que es lo mismo, no podrá realizarse amplificación alguna si no se tiene al menos una molécula completa del ADN a amplificar. El sesgo producido en la distribución gaussiana del error puede provocar esta aparición de datos extraños o «amortiguaciones medias».
Luego la recta de calibrado permite una cuantificación para un protocolo experimental dado pero hay que tener en cuenta que existen multitud de fuentes de error potenciales, como diferencias de composición química del tampón de reacción, de las muestras (presencia de proteínas, ARN, etc.) e incluso del diluyente (el agua, generalmente).
Enfoques
Como se indicaba anteriormente, es posible cuantificar la expresión génica tanto en términos absolutos como en relativos (es decir, por comparación con la expresión de otro gen). En el segundo caso, es de vital importancia seleccionar como gen estándar aquel cuya expresión realmente no varíe cuando se somete al individuo al tratamiento experimental cuyo efecto en la transcripción se desea estudiar (por ejemplo, un estrés ambiental,[17] biótico[18] o tipo de tejido[19]).
- Cuantificación absoluta
Se basa en las características de la fase exponencial de la curva sigmoide de emisión de fluorescencia.[20] Responde a la ecuación:

donde Q es la cantidad de ADN, n corresponde al número de ciclo, o es el ciclo de partida y E la eficiencia de la reacción.
En el ciclo en el que se sobrepasa el nivel umbral (esto es, el CT), se dice que:
(1)

- Cuantificación relativa
Establece una relación R entre la cantidad de ADN inicial de una muestra respecto de la de un testigo, que se asume que se expresa de forma invariable e independiente del tratamiento. En su detección tenemos que:[4]

- sustituyendo de (1) tenemos que

- y, por tanto
o:


Desde 2002 existe una tendencia a emplear varios genes normalizadores para realizar una cuantificación relativa fiable; de este modo, se generan factores de normalización que ponderan de algún modo el impacto de cada gen interno; una aproximación común se basa en el empleo de medias geométricas de estos genes normalizadores.[15]
Selección de genes de normalización
El análisis estadístico que permite la detección de los genes expresados de forma más estable en la serie de tejidos estudiada es crucial, puesto que de él depende la elección de los genes que se emplearán como control interno durante los ensayos de cuantificación relativa. Con el fin de evaluar esta estabilidad, en algunos casos se comparan sus valores CT; en otros, sus cantidades relativas empleando la muestra cuyo CT es menor como elemento normalizador.
Como primera aproximación, el algoritmo geNorm[15] permite el cálculo de un valor de estabilidad M inversamente proporcional a la estabilidad del gen en cuestión, y de una serie de valores "PV" que definen el número mínimo de genes adecuado para efectuar una normalización fiable. El cómputo de valor M se basa en definir la variación media, calculada comparando pares dos a dos, de un gen frente a todos los demás genes presentes en el estudio. El algoritmo se realiza en varios pasos, ya que en cada ronda de comparaciones se suprime el gen que ha obtenido un peor valor; de este modo, es capaz de adjudicar un valor M a cada gen salvo para los más estables, que comparten el mismo. En cuanto a valores PV, su cómputo se basa en el diseño de un factor de normalización óptimo; al principio, este se realiza con los dos genes más estables pero, secuencialmente, se le van añadiendo otros hasta que dicha adición no mejora significativamente la precisión de aquel. Así, se define un valor PV para las comparaciones entre Vn/n+1, estimado como la desviación estándar de las ratios transformadas logarítmicamente de .

Otro algoritmo, denominado NormFinder, aprovecha los valores CT para ajustar un modelo matemático de la expresión génica capaz de evaluar las variaciones presentes dentro y entre grupos (tipo de órganos, tratamientos químicos... ) definidos por el investigador. De este modo, los genes con una variación inter e intragrupo mínima son los considerados más estables, con un valor de estabilidad S mínimo.[21]
También es posible evaluar no ya la estabilidad mediante comparaciones dos a dos, sino analizar el coeficiente de variación de los niveles de expresión relativos normalizados. Esta aproximación, propia de qBase o qBasePlus,[22] requiere la transformación de los valores CT iniciales en cantidades relativas, teniendo en cuenta los valores de eficiencia de la PCR y empleando como calibrador el gen con menor CT; tras ello, se calcula un factor de normalización para cada muestra empleando la media geométrica de los valores relativos estimados para todos los genes candidatos.
Controles
Se deben realizar varios tipos de controles:
- Control negativo: añadimos todos los reactivos necesarios menos la muestra, por lo que no deberemos obtener amplificación
- Control de inhibidores (control positivo):
- Externos: realizaremos un tubo en paralelo para cada muestra en el que se amplifica un fragmento a partir de una cantidad conocida de copias (mismos cebadores, con deleción o inserción u otro fragmento no relacionado)
- Internos: Se añade una cantidad mínima conocida y detectable de una secuencia modificada de la secuencia diana que se amplifica, con los mismos cebadores pero que se detecta con un fluorocromo distinto.
- Control de cuantificación (estándares):
- Externos: se utiliza la misma secuencia que se quiere cuantificar pero modificando, bien en el sitio de la sonda, bien en otro lugar. Esto evita problemas de contaminación.
- Control sin transcripción reversa: este se utiliza cuando el templado es un cADN preparado a partir de un paso de transcripción reversa desde un ARN. Este control es especialmente útil para evaluar el paso de purificación de los ARN y evitar contaminantes.
Empleo de sondas específicas
La PCR en tiempo real puede realizarse marcando de manera fluorescente oligonucleótidos que detectan específicamente la aparición del producto deseado. El fundamento de esta técnica se basa en el empleo del FRET o transferencia de energía de resonancia de Förster, que es un mecanismo de transferencia de energía entre cromóforos. EL FRET se fundamenta en que la excitación de un cromóforo puede transferirse a otro cercano, generalmente cuando ambos se sitúan en la misma molécula, mediante un mecanismo acoplador dipolo-dipolo.[23] En el caso de que los cromóforos sean fluorescentes (esto es, fluorocromos), el mecanismo subyacente continúa siendo el mismo: la energía se transfiere, lo que desemboca en la aparición de fluorescencia (cabe destacar que no es la fluorescencia la transferida)[24][25] Las sondas empleadas en PCR a tiempo real existentes son:
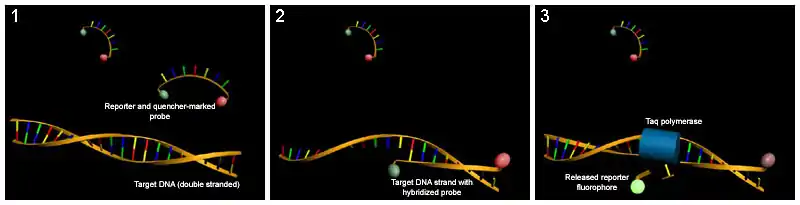
- Las sondas TaqMan permiten medir la producción de productos de PCR mediante un sistema de sondas marcadas mediante dos fluorocromos. Su utilidad radica en que poseen un fluoróforo en su extremo 3' y una molécula en el 5' que bloquea su emisión de fluorescencia (denominada en inglés «quencher»); esta sonda marcada hibrida específicamente en la parte central del producto de PCR a obtener. De este modo, cuando se efectúa la PCR (con la sonda más el par de cebadores específicos), la sonda híbrida en el amplicón, pero, debido a la cercanía del fluoróforo al quencher, no se emite fluorescencia. Cuando la polimerasa se topa con la sonda, la hidroliza mediante su actividad exonucleasa 5'-3', lo cual provoca la separación del quencher del fluorocromo y, por tanto, la emisión de fluorescencia, que está relacionada con la cantidad de amplicón producido.[26] Hay que resaltar dos ventajas primordiales frente a las sondas inespecíficas, como el bromuro de etidio o el SYBR GREEN: con las sondas TaqMan la fluorescencia que se produce es específica de la amplificación que estemos estudiando, además de permitir que se usen varios fluorocromos en la misma reacción y detectar varios ADN/ ARN al mismo tiempo. La desventaja con la que nos encontramos es que hay que diseñar sondas específicas en cada estudio, mientras que con las inespecíficas no se necesitaría diseñar una nueva para cada ensayo lo que, por otro lado, provoca que aumente el número de falsos positivos (disminuye especificidad)
- Las sondas tipo Molecular Beacons son también oligonucleótidos de cadena simple que, por su estructura, poseen una zona de apareamiento de bases interna y que, por tanto, forman una horquilla. En presencia del amplicón la sonda se abre y se une preferentemente a aquel, lo que produce la emisión de fluorescencia. Estructuralmente, poseen la zona complementaria al amplicón en el giro de la horquilla, la de complementariedad interna en el cuello y los fluoróforos en sus extremos: el donador en el 5' y el aceptor en el 3'. Cuando la sonda está cerrada en horquilla, el fluoróforo del 3' impide la emisión de fluorescencia por parte del propio del 5', cosa que no sucede al unirse al amplicón.[27]
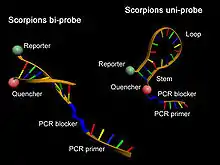
- Sondas Scorpion. Se trata de moléculas mixtas que contienen un cebador específico para el amplicón unido covalentemente a una horquilla similar a las sondas del tipo Molecular Beacon que, en su zona de giro, poseen un elemento complementario al amplicón. Los fluoróforos donador y aceptor se encuentran en la estructura de horquilla; de este modo, cuando esta se encuentra cerrada no se produce la emisión de fluorescencia, lo que sí sucede cuando ambos se separan debido a la presencia del amplicón y la apertura de la zona en horquilla.[28]
Aplicaciones tecnológicas
Si bien el uso más ampliamente difundido de la Q-PCR consiste en la evaluación de la expresión génica de genes concretos de forma relativa (empleando ARNm de la muestra y retrotranscribiéndolo a ADNc, que se mide mediante esta técnica), se han desarrollado otras aplicaciones fuera del entorno puramente académico. Las aplicaciones de impacto en la industria incluyen la cuantificación de carga microbiana en alimentos o en material vegetal, la detección de OGM (organismos genéticamente modificados) y la cuantificación y genotipado de patógenos virales en humanos.
Usos en diagnóstico
La PCR cuantitativa se utiliza para el diagnóstico de enfermedades, tales como enfermedades infecciosas, cáncer y anormalidades genéticas. La introducción de ensayos de PCR cuantitativa en laboratorios de microbiología ha mejorado el diagnóstico de enfermedades infecciosas y se utiliza además como herramienta para detectar nuevas enfermedades emergentes.
Usos microbiológicos
La Q-PCR es utilizada también por microbiólogos en el campo de seguridad y deterioro de los alimentos, riesgo microbiológico en la calidad del agua y en la protección de la salud pública.
Usos en investigación
En el campo de la investigación, la PCR cuantitativa se utiliza mayormente para mediciones cuantitativas de la transcripción y expresión génica, determinando como cambia la expresión de un gen concreto a lo largo del tiempo o en determinadas condiciones como la administración de un fármaco, y analizando la respuesta de las células de un tejido al mismo, además del progreso de la diferenciación celular o respuestas frente a cambios en las condiciones ambientales. También se utiliza para determinar la cigosidad de animales transgénicos utilizados en investigación, es decir, determinar la homocigosis o heterocigosis de un individuo para un determinado gen.
Detección de fitopatógenos
La producción de propágulos vegetales o plántulas libres de patógenos es una constante en la industria agronómica por cuestiones económicas y sanitarias. Para el caso de Phytophthora ramorum, un hongo que causa muerte súbita en robles y otras especies, se han desarrollado sistemas basados en sondas Taqman capaces de detectar una cantidad de 10 a 100 fg mezclados en el ADN de la planta hospedadora. La discriminación entre el ADN del patógeno y de la planta se hace amplificando secuencias espaciadoras transcritas internas (ITS), unos espaciadores situados en la zona codificante de los genes de ARN ribosomal característicos para cada taxón.[29] Existen variantes con sondas Scorpion, Molecular Beacon y LAMP para el mismo patógeno que, además, pueden ser aplicadas en campo.[30]
Detección de OGM
La Q-PCR (precedida de retrotranscripción) se emplea en la detección de OGM debido a la alta sensibilidad y rango dinámico de detección de ADN; sus alternativas, como son el análisis del ADN o de las proteínas, suelen mostrar menos sensibilidad. Para ello se emplean cebadores específicos que amplifiquen no ya el propio transgén, sino su promotor, terminador e incluso sus secuencias intermedias, empleadas durante el proceso de ingeniería del vector. Además, puesto que el proceso de creación de una planta transgénica suele conducir a la inserción de más de una copia del transgén, debe evaluarse su cantidad; para ello se efectúa una cuantificación relativa empleando como gen control uno propio de la especie tratada, que se encuentre en copia única.[31][32]
Cuantificación y genotipado en clínica
Las virosis en humanos pueden ser debidas a infecciones por parte de patógenos concretos o a coinfecciones, y este hecho no solo puede dificultar el diagnóstico mediante las técnicas clásicas, sino que puede redundar en un pronóstico diferente y en la necesidad de emplear una u otra quimioterapia. El empleo de la Q-PCR posibilita tanto la cuantificación como el genotipado (caracterización de la cepa, que se hace mediante curvas de melting) de virus como el HBV (virus de la hepatitis B).[33] El grado de infección, cuantificado como las copias de genoma viral por unidad de tejido del paciente, es relevante en muchos casos. Por ejemplo, la probabilidad de que el virus del herpes simple de tipo 1 se reactive está relacionada con el número de neuronas infectadas en los ganglios.[34] Esta cuantificación se realiza con retrotranscripción o sin ella, en el caso de que los virus se integren en el genoma humano en algún momento de su ciclo, como en el caso del HPV (virus del papiloma humano), alguna de cuyas variantes está asociada con la aparición de cáncer de cérvix.[35]
Referencias
- Watson, J. D.; Baker, T. A.; Bell, S. P.; Gann, A.; Levine, M. y Losick, R. (2004). Molecular Biology of the Gene (Fifth edition edición). San Francisco: Benjamin Cummings. ISBN 0-321-22368-3.
- Pfaff, Michael W., Ales Tichopad, Christian Prgomet y Tanja P. Neuvians (2005). «Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper – Excel-based tool using pair-wise correlations.» Archivado el 13 de septiembre de 2019 en Wayback Machine. Biotechonology Letters 26:509-515.
- Vandesompele, J., De Preter, K., Pattyn, F., Poppe, B., Van Roy, N., De Paepe, A., Speleman, F. (2002) «Accurate normalisation of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes.» Archivado el 27 de mayo de 2020 en Wayback Machine. Gen. Biol. 3: 1-12.
- Pfaffl, M. W., Horgan, G. W., Dempfle, L. (2002) «Relative Expression Software Tool (REST©) for group wise comparison and statistical analysis of relative expression results in real-time PCR.» Nucl. Acids Res. 30: e36.
- Rychlik, W., Spencer, W. J., Rhoads, R. E. (1990). «Optimization of the annealing temperature for DNA amplification in vitro». Nucl Acids Res 18: 6409-6412. PMID 2243783. doi:10.1093/nar/18.21.6409.
- Sambrook, Joseph y David W. Russel (2001). Molecular Cloning: A Laboratory Manual (3rd ed. edición). Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press. ISBN 0-87969-576-5.
- Zipper, H. et al. (2004). «Investigations on DNA intercalation and surface binding by SYBR Green I, its structure determination and methodological implications.» Nucleic Acids Res. 32, e103. PMID 15249599
- Ponchel, F.; Toomes, C.; Bransfield, K.; Leong, F. T.; Douglas, S. H.; Field, S. L.; Bell, S. M.; Combaret, V.; Puisieux, A.; Mighell, A. J.; Others (2003), «… for a relative quantification of gene rearrangements, gene amplifications and micro gene deletions» (w), BMC Biotechnol 3 (1): 18, doi:10.1186/1472-6750-3-18.
- Ririe, K. M.; Rasmussen, R. P.; Wittwer, C. T. (1997), «Product Differentiation by Analysis of DNA Melting Curves during the Polymerase Chain Reaction», Analytical Biochemistry 245 (2): 154-160, doi:10.1006/abio.1996.9916, archivado desde el original el 5 de agosto de 2004, consultado el 23 de noviembre de 2008.
- Brunner, A. M., Yakovlev, I. A., Strauss, S. H. (2004) Validating internal controls for quantitative plant gene expression studies. BMC Plant Biol 4: 14
- Czechowski, T., Stitt, M., Altmann, T., Udvardi, M. K., Scheible, W. R. (2005) Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis (enlace roto disponible en Internet Archive; véase el historial, la primera versión y la última).. Plant Physiol 139: 5-17
- Thellin, O., Zorzi, W., Lakaye, B., De Borman, B., Coumans, B., Henne, G., Grisar, T., Igout, A., Heinen, E. (1999) «Housekeeping genes as internal standards: use and limits.» J Biotechnol 75: 197-200
- Radonic, A., Thulke, S., Mackay, I. M., Landt, O., Siegert, W., Nitsche, A. (2004) Guideline for reference gene selection for quantitative real-time PCR. Biochem Biophys Res Commun 313: 856-862
- Dheda, K., Huggett, J. F., Bustin, S. A., Johnson, M. A., Rook, G., Zumla, A. (2004) Validation of housekeeping genes for normalizing RNA expression in real-time PCR. Biotechniques 37: 112-119
- Vandesompele, J., De Preter, K., Pattyn, F., Poppe, B., Van Roy, N., De Paepe, A., Speleman, F. (2002) «Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes.» Genome Biol 37: RESEARCH0034
- Pfaffl, M. W., Tichopad, A., Prgomet, C., Neuvians, T. P. (2004) Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper—Excel-based tool using pair-wise correlations. Biotechnol Lett 26: 509-515
- .
- Nicot, Nathalie; Hausman, Jean-Francois; Hoffmann, Lucien; Evers, Daniele (2005), «Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress», Journal of Experimental Botany 56 (421): 2907-2914, PMID 16188960, doi:10.1093/jxb/eri285.
- Dheda, K.; Huggett, J. F.; Bustin, S. A.; Johnson, M. A.; Rook, G.; Zumla, A. (2004), Biotechniques 37: 112-119 http://www.gene-quantification.de/dheda-bustin-hkg-2004.pdf
|url=sin título (ayuda). - Bustin, S. A. (2000), «Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays», Journal of Molecular Endocrinology 25 (2): 169-193, PMID 11013345, doi:10.1677/jme.0.0250169, archivado desde el original el 6 de julio de 2010, consultado el 20 de diciembre de 2008.
- Andersen, Claus Lindbjerg; Jensen, Jens Ledet; Orntoft, Torben Falck (2004), «Normalization of Real-Time Quantitative Reverse Transcription-PCR Data: A Model-Based Variance Estimation Approach to Identify Genes Suited for Normalization, Applied to Bladder and Colon Cancer Data Sets» (w), Cancer Research 64 (15): 5245-5250, PMID 15289330, doi:10.1158/0008-5472.CAN-04-0496.
- Hellemans, J.; Mortier, G.; De Paepe, A.; Speleman, F.; Vandesompele, J. (2007), «qBase relative quantification framework and software for management and automated analysis of real- …», Genome Biol 8 (2): R19, doi:10.1186/gb-2007-8-2-r19, archivado desde el original el 19 de abril de 2015, consultado el 5 de febrero de 2009.
- Förster, T., Zwischenmolekulare Energiewanderung und Fluoreszenz, Ann. Physik 1948, 437, 55. doi 10.1002/andp.19484370105
- Lakowicz, Joseph R. Principles of Fluorescence Spectroscopy. Plenum Publishing Corporation, 2nd edition (July 1, 1999)
- FRET microscopy tutorial from Olympus
- Heid, C. A.; Stevens, J.; Livak, K. J.; Williams, P. M. (1996), «Real time quantitative PCR», Genome Research 6 (10): 986-994, doi:10.1101/gr.6.10.986.
- Tyagi, S.; Kramer, F. R.; y otros (1996), «Molecular Beacons: Probes that Fluoresce upon Hybridization», Nature Biotechnology 14 (3): 303-308, doi:10.1038/nbt0396-303, archivado desde el original el 7 de septiembre de 2006, consultado el 20 de diciembre de 2008.
- Whitcombe, D.; Theaker, J.; Guy, S. P.; Brown, T.; Little, S. (1999), Nature Biotechnology 17: 804-807, doi:10.1038/11751 http://www.gene-quantification.de/whitcombe-1999.pdf
|url=sin título (ayuda). - Baldwin, B. G. (1992). «Phylogenetic utility of the internal transcribed spacers of nuclear ribosomal DNA in plants: An example from the Compositaogy». Molecular Phylogenetics and Evolution 1: 3-16. doi:10.1016/1055-7903(92)90030-K.
- Tomlinson, J. A.; Barker, I.; Boonham, N. (2007), «Faster, Simpler, More-Specific Methods for Improved Molecular Detection of Phytophthora ramorum in the Field» (w), Applied and Environmental Microbiology 73 (12): 4040-4047, PMID 17449689, doi:10.1128/AEM.00161-07.
- Holst-Jensen, A.; Ronning, S. B.; Lovseth, A.; Berdal, K. G. (2003), «PCR technology for screening and quantification of genetically modified organisms (GMOs)» (w), Analytical and Bioanalytical Chemistry 375 (8): 985-993.
- Brodmann, P. D.; Ilg, E. C.; Berthoud, H.; Herrmann, A. (2002), «… -Time Quantitative Polymerase Chain Reaction Methods for Four Genetically Modified Maize Varieties …», Journal of AOAC International 85 (3): 646-653, doi:10.5555/jaoi.2002.85.3.646.
- Yeh, S. H.; Tsai, C. Y.; Kao, J. H.; Liu, C. J.; Kuo, T. J.; Lin, M. W.; Huang, W. L.; Lu, S. F.; Jih, J.; Chen, D. S.; y otros (2004), «Quantification and genotyping of hepatitis B virus in a single reaction by real-time PCR and melting …», Journal of Hepatology 41 (4): 659-666, doi:10.1016/j.jhep.2004.06.031.
- Sawtell, N. M. (1998), «The Probability of in Vivo Reactivation of Herpes Simplex Virus Type 1 Increases with the Number of Latently Infected Neurons in the Ganglia», Journal of Virology 72 (8): 6888-6892.
- Peter, M.; Rosty, C.; Couturier, J.; Radvanyi, F.; Teshima, H.; Sastre-garau, X. (2006), «MYC activation associated with the integration of HPV DNA at the MYC locus in genital tumors» (w), Oncogene 25 (44): 5985-5993, doi:10.1038/sj.onc.1209625.
Bibliografía
- Poitras, Elyse y Alain Houde (2002). «La PCR en temps réel: principes et applications.» Reviews in Biology and Biotechnology. 2(2):2-11.
- Bustin, S. A. (2000). «Absolute quantification of m ARN using real-time reverse transcription polymerase chain reaction assays.» J Mol Endocrinol. 25(2):169-93.
- Higuchi, R., Dollinger, G., Walsh, P. S., Griffith, R. (1992). «Simultaneous amplification and detection of specific ADN-sequences.» Bio-Technology 10 (4), 413-417.
- Holland, P. M., Abramson, R. D., Watson, R., Gelfand, D. H. (1991). «Detection of specific polymerase chain reaction product by utilizing the 50 !30 exonuclease activity of Thermus aquaticus ADN polymerase.» Proc. Natl. Acad. Sci. USA 88 (16), 7276-7280.
- Kubista, M., Andrade, J. M., Bengtsson, M., Forootan, A., Jonak, J., Lind, K., Sindelka, R., Sjoback, R., Sjogreen, B., Strombom, L., Stahlberg, A., Zoric, N. (2006). «The real-time polymerase chain reaction.» Aspects Med. 27(2-3):95-125.
Enlaces externos
- The Reference in Q-PCR Academic & Industrial Information Platform (en inglés)
- Q-PCR tutorial. University of South Carolina (en inglés)
- Articles about Real Time Pcr (en inglés)
| Control de autoridades |
|
|---|
 Datos: Q856198
Datos: Q856198 Multimedia: Real-time polymerase chain reaction / Q856198
Multimedia: Real-time polymerase chain reaction / Q856198