Regulación transcripcional
En biología molecular y genética, la regulación transcripcional es el medio por el cual una célula regula la conversión de ADN en ARN (transcripción), orquestando así la actividad genética. Un solo gen puede regularse de diversas formas, desde la alteración del número de copias de ARN que se transcriben hasta el control temporal de cuándo se transcribe el gen. Este control permite que la célula o el organismo responda a una variedad de señales intra y extracelulares y, por lo tanto, genere una respuesta. Algunos ejemplos de esto incluyen producir el ARNm que codifica enzimas para adaptarse a un cambio en una fuente de alimento, producir los productos génicos involucrados en actividades específicas del ciclo celular y producir los productos génicos responsables de la diferenciación celular en eucariotas multicelulares, como se estudió en la biología evolutiva del desarrollo evolutivo.
| Glosario de regulación de la transcripción |
|---|
| • regulación transcripcional: controla la tasa de transcripción de genes, por ejemplo, ayudando o dificultando la unión de la ARN polimerasa al ADN. |
| • transcripción: el proceso de fabricación de ARN a partir de una plantilla de ADN mediante la ARN polimerasa |
| • factor de transcripción: una sustancia, como una proteína, que contribuye a la causa de una reacción bioquímica o proceso corporal específico |
| • promotor: una región del ADN que inicia la transcripción de un gen en particular |
| • factor sigma: cofactores bacterianos especializados que forman complejos con la ARN polimerasa y codifican la especificidad de la secuencia |
| • coactivador: una proteína que trabaja con factores de transcripción para aumentar la tasa de transcripción de genes |
| • correpresor: una proteína que trabaja con factores de transcripción para disminuir la tasa de transcripción de genes |
La regulación de la transcripción es un proceso vital en todos los organismos vivos. Está orquestado por factores de transcripción y otras proteínas que trabajan en conjunto para ajustar con precisión la cantidad de ARN que se produce a través de una variedad de mecanismos. Las bacterias y los eucariotas tienen estrategias muy diferentes para lograr el control sobre la transcripción, pero algunas características importantes permanecen conservadas entre las dos. Lo más importante es la idea del control combinatorio, que es que cualquier gen dado probablemente esté controlado por una combinación específica de factores para controlar la transcripción. En un ejemplo hipotético, los factores A y B podrían regular un conjunto distinto de genes de la combinación de los factores A y C. Esta naturaleza combinatoria se extiende a complejos de mucho más de dos proteínas y permite que un subconjunto muy pequeño (menos del 10%) del genoma para controlar el programa transcripcional de toda la célula.
En bacterias
Gran parte de la comprensión temprana de la transcripción provino de bacterias,[1] aunque el alcance y la complejidad de la regulación transcripcional es mayor en eucariotas. La transcripción bacteriana se rige por tres elementos principales de secuencia:
- Los promotores son elementos del ADN que pueden unirse a la ARN polimerasa y otras proteínas para el inicio exitoso de la transcripción directamente aguas arriba del gen.
- Los operadores reconocen proteínas represoras que se unen a un tramo de ADN e inhiben la transcripción del gen.
- Elementos de control positivo que se unen al ADN e incitan a niveles más altos de transcripción.[2]
Si bien estos medios de regulación transcripcional también existen en eucariotas, el panorama transcripcional es significativamente más complicado tanto por el número de proteínas involucradas como por la presencia de intrones y el empaquetamiento del ADN en histonas.
La transcripción de un gen bacteriano básico depende de la fuerza de su promotor y de la presencia de activadores o represores. En ausencia de otros elementos reguladores, la afinidad basada en la secuencia de un promotor por las ARN polimerasas varía, lo que da como resultado la producción de diferentes cantidades de transcripción. La afinidad variable de la ARN polimerasa por diferentes secuencias promotoras está relacionada con las regiones de la secuencia consenso cadena arriba del sitio de inicio de la transcripción. Cuantos más nucleótidos de un promotor coincidan con la secuencia de consenso, más probable será la afinidad del promotor por la ARN polimerasa.[3]
En ausencia de otros elementos reguladores, el estado predeterminado de una transcripción bacteriana es estar en la configuración "activada", lo que resulta en la producción de cierta cantidad de transcripción. Esto significa que la regulación transcripcional en forma de represores de proteínas y elementos de control positivo puede aumentar o disminuir la transcripción. Los represores a menudo ocupan físicamente la ubicación del promotor, impidiendo la unión de la ARN polimerasa. Alternativamente, un represor y una polimerasa pueden unirse al ADN al mismo tiempo con una interacción física entre el represor que impide la apertura del ADN para acceder a la hebra negativa para la transcripción. Esta estrategia de control es distinta de la transcripción eucariota, cuyo estado basal debe estar apagado y donde los cofactores necesarios para el inicio de la transcripción son altamente dependientes de genes.[4]
Los factores sigma son proteínas bacterianas especializadas que se unen a las ARN polimerasas y orquestan el inicio de la transcripción. Los factores sigma actúan como mediadores de la transcripción específica de secuencia, de modo que se puede usar un solo factor sigma para la transcripción de todos los genes domésticos o un conjunto de genes que la célula desea expresar en respuesta a algunos estímulos externos como el estrés.[5]

Además de los procesos que regulan la transcripción en la etapa de inicio, la síntesis de ARNm también está controlada por la tasa de elongación de la transcripción.[7] Las pausas de la ARN polimerasa ocurren con frecuencia y están reguladas por factores de transcripción, como NusG y NusA, acoplamiento transcripción-traducción y estructura secundaria del ARNm.[8][9]
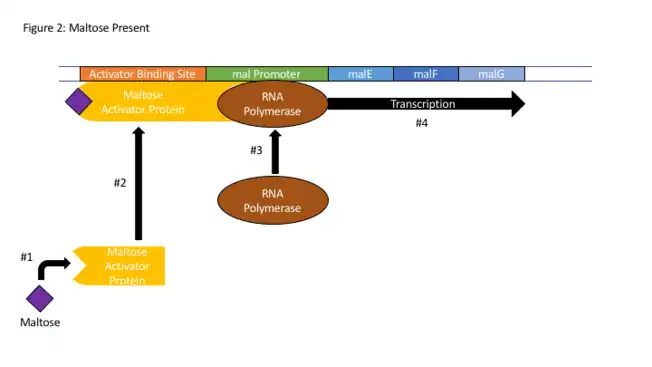
En eucariotas
La complejidad adicional de generar una célula eucariota conlleva un aumento en la complejidad de la regulación transcripcional. Los eucariotas tienen tres ARN polimerasas, conocidas como Pol I, Pol II y Pol III. Cada polimerasa tiene objetivos y actividades específicos y está regulada por mecanismos independientes. Hay varios mecanismos adicionales a través de los cuales se puede controlar la actividad de la polimerasa. Estos mecanismos se pueden agrupar generalmente en tres áreas principales:
- Control sobre el acceso de la polimerasa al gen. Este es quizás el más amplio de los tres mecanismos de control. Esto incluye las funciones de las enzimas remodeladoras de histonas, factores de transcripción, potenciadores y represores, y muchos otros complejos.
- Alargamiento productivo de la transcripción de ARN. Una vez que la polimerasa se une a un promotor, requiere otro conjunto de factores para permitirle escapar del complejo del promotor y comenzar a transcribir con éxito el ARN.
- Terminación de la polimerasa. Se ha descubierto que varios factores controlan cómo y cuándo se produce la terminación, lo que determinará el destino de la transcripción de ARN.
Los tres de estos sistemas funcionan en conjunto para integrar señales de la célula y cambiar el programa transcripcional en consecuencia.
Mientras que en los sistemas procariotas el estado de transcripción basal puede considerarse no restrictivo (es decir, "activado" en ausencia de factores modificadores), los eucariotas tienen un estado basal restrictivo que requiere el reclutamiento de otros factores para generar transcripciones de ARN. Esta diferencia se debe en gran parte a la compactación del genoma eucariota al enrollar el ADN alrededor de las histonas para formar estructuras de orden superior. Esta compactación hace que el promotor del gen sea inaccesible sin la ayuda de otros factores en el núcleo y, por tanto, la estructura de la cromatina es un sitio común de regulación. De manera similar a los factores sigma en procariotas, los factores de transcripción general (GTF) son un conjunto de factores en eucariotas que son necesarios para todos los eventos de transcripción. Estos factores son responsables de estabilizar las interacciones de unión y abrir la hélice de ADN para permitir que la ARN polimerasa acceda a la plantilla, pero generalmente carecen de especificidad para diferentes sitios promotores.[13] Una gran parte de la regulación génica se produce a través de factores de transcripción que reclutan o inhiben la unión de la maquinaria de transcripción general y/o la polimerasa. Esto se puede lograr a través de interacciones cercanas con los elementos promotores centrales o mediante los elementos potenciadores de larga distancia.
Una vez que una polimerasa se une con éxito a una plantilla de ADN, a menudo requiere la ayuda de otras proteínas para dejar el complejo promotor estable y comenzar a alargar la cadena de ARN naciente. Este proceso se denomina escape del promotor y es otro paso en el que los elementos reguladores pueden actuar para acelerar o ralentizar el proceso de transcripción. De manera similar, los factores de proteínas y ácidos nucleicos pueden asociarse con el complejo de elongación y modular la velocidad a la que la polimerasa se mueve a lo largo de la plantilla de ADN.
A nivel de estado de cromatina
En eucariotas, el ADN genómico está muy compactado para poder encajarlo en el núcleo. Esto se logra enrollando el ADN alrededor de octámeros de proteínas llamados histonas, lo que tiene consecuencias para la accesibilidad física de partes del genoma en un momento dado. Porciones significativas se silencian mediante modificaciones de histonas y, por lo tanto, son inaccesibles para las polimerasas o sus cofactores. El nivel más alto de regulación de la transcripción ocurre a través del reordenamiento de histonas para exponer o secuestrar genes, porque estos procesos tienen la capacidad de hacer inaccesibles regiones enteras de un cromosoma, como ocurre en la impronta.
El reordenamiento de las histonas se facilita mediante modificaciones postraduccionales de las colas de las histonas centrales. Se pueden realizar una amplia variedad de modificaciones mediante enzimas como las histonas acetiltransferasas (HAT), histonas metiltransferasas (HMT) e histonas desacetilasas (HDAC), entre otras. Estas enzimas pueden agregar o eliminar modificaciones covalentes como grupos metilo, grupos acetilo, fosfatos y ubiquitina. Las modificaciones de histonas sirven para reclutar otras proteínas que pueden aumentar la compactación de la cromatina y secuestrar elementos promotores, o aumentar el espaciamiento entre histonas y permitir la asociación de factores de transcripción o polimerasa en ADN abierto.[14] Por ejemplo, la trimetilación de H3K27 por el complejo polycomb PRC2 provoca la compactación cromosómica y el silenciamiento de genes.[15] Estas modificaciones de histonas pueden ser creadas por la célula o heredadas de forma epigenética de un padre.
A nivel de metilación de citosina

La 5-metilcitosina es una forma metilada de la citosina [16] (C) de la base del ADN que regula la transcripción de genes en mamíferos.[17] Las citosinas metiladas se encuentran principalmente en secuencias de dinucleótidos donde la citosina es seguida por una guanina, un sitio CpG. El número total de sitios CpG en el genoma humano es de aproximadamente 28 millones[18] y generalmente alrededor del 70% de todos los sitios CpG tienen una citosina metilada.[19] La metilación de CpG en una región promotora de un gen reprime la transcripción[20] mientras que la metilación de CpG en el cuerpo de un gen aumenta la expresión.[21] Las enzimas TET juegan un papel central en la desmetilación de citosinas metiladas. La desmetilación de CpG en un promotor de gen mediante la actividad de la enzima TET aumenta la transcripción del gen.[16]
Factores de transcripción
Los factores de transcripción son proteínas que se unen a secuencias de ADN específicas para regular la expresión de un gen determinado. Hay aproximadamente 1.400 factores de transcripción en el genoma humano y constituyen aproximadamente el 6% de todos los genes codificadores de proteínas humanas.[22] El poder de los factores de transcripción reside en su capacidad para activar y/o reprimir amplios repertorios de genes diana aguas abajo. El hecho de que estos factores de transcripción funcionen de manera combinatoria significa que solo un pequeño subconjunto del genoma de un organismo codifica factores de transcripción. Los factores de transcripción funcionan a través de una amplia variedad de mecanismos. En un mecanismo, la metilación de CpG influye en la unión de la mayoría de los factores de transcripción al ADN, en algunos casos de forma negativa y en otros de forma positiva.[23] Además, a menudo se encuentran al final de una vía de transducción de señales que funciona para cambiar algo sobre el factor, como su localización subcelular o su actividad. Las modificaciones postraduccionales de los factores de transcripción ubicados en el citosol pueden hacer que se trasladen al núcleo donde pueden interactuar con sus potenciadores correspondientes. Otros factores de transcripción ya están en el núcleo y se modifican para permitir la interacción con factores de transcripción asociados. Algunas modificaciones post-traduccionales conocidas para regular el estado funcional de los factores de transcripción son fosforilación, acetilación, SUMOlación y ubiquitilación. Los factores de transcripción se pueden dividir en dos categorías principales: activadores y represores. Mientras que los activadores pueden interactuar directa o indirectamente con la maquinaria central de la transcripción a través de la unión del potenciador, los represores reclutan predominantemente complejos correpresores que conducen a la represión transcripcional por la condensación de cromatina de las regiones potenciadoras. También puede suceder que un represor funcione mediante competencia alostérica contra un activador determinado para reprimir la expresión génica: los motivos de unión al ADN superpuestos tanto para los activadores como para los represores inducen una competencia física para ocupar el sitio de unión. Si el represor tiene una mayor afinidad por su motivo que el activador, la transcripción se bloquearía eficazmente en presencia del represor. El estricto control regulador se logra mediante la naturaleza altamente dinámica de los factores de transcripción. Nuevamente, existen muchos mecanismos diferentes para controlar si un factor de transcripción está activo. Estos mecanismos incluyen el control sobre la localización de la proteína o el control sobre si la proteína puede unirse al ADN.[24] Un ejemplo de esto es la proteína HSF1, que permanece unida a Hsp70 en el citosol y solo se transloca al núcleo ante estrés celular como el choque térmico. Por tanto, los genes bajo el control de este factor de transcripción permanecerán sin transcribir a menos que la célula esté sometida a estrés.[25]
Potenciadores
Los potenciadores o módulos/elementos reguladores cis (CRM/CRE) son secuencias de ADN no codificantes que contienen múltiples sitios de unión de activadores y represores. Los potenciadores varían de 200 pb a 1 kb de longitud y pueden ser proximales, 5' aguas arriba del promotor o dentro del primer intrón del gen regulado, o distales, en intrones de genes vecinos o regiones intergénicas alejadas del locus. A través del bucle de ADN, los potenciadores activos se ponen en contacto con el promotor de forma dependiente de la especificidad del promotor del motivo de unión al ADN del núcleo.[26] La dicotomía promotor-potenciador proporciona la base para la interacción funcional entre los factores de transcripción y la maquinaria del núcleo de la transcripción para desencadenar el escape del RNA Pol II del promotor. Mientras que uno podría pensar que existe una relación potenciador-promotor de 1:1, los estudios del genoma humano predicen que un promotor activo interactúa con 4 a 5 potenciadores. De manera similar, los potenciadores pueden regular más de un gen sin restricción de ligamiento y se dice que "saltan" genes vecinos para regular los más distantes. Aunque es poco frecuente, la regulación transcripcional puede involucrar elementos ubicados en un cromosoma diferente al que reside el promotor. Los potenciadores o promotores proximales de genes vecinos pueden servir como plataformas para reclutar elementos más distales.[27]
Panorama regulatorio
La iniciación, terminación y regulación de la transcripción están mediadas por el "bucle de ADN" que reúne promotores, potenciadores, factores de transcripción y factores de procesamiento de ARN para regular con precisión la expresión génica.[28] La captura de conformación cromosómica (3C) y, más recientemente, las técnicas de Hi-C proporcionaron evidencia de que las regiones de cromatina activa están "compactadas" en dominios o cuerpos nucleares donde se mejora la regulación transcripcional.[29] La configuración del genoma es fundamental para la proximidad potenciador-promotor. Las decisiones sobre el destino celular están mediadas por reorganizaciones genómicas altamente dinámicas en la interfase para activar o desactivar de forma modular redes reguladoras de genes completas a través de reordenamientos de cromatina de corto a largo alcance.[30] Estudios relacionados demuestran que los genomas de los metazoos se dividen en unidades estructurales y funcionales alrededor de una megabase larga llamada dominios de asociación topológica (TAD) que contiene docenas de genes regulados por cientos de potenciadores distribuidos dentro de grandes regiones genómicas que contienen solo secuencias no codificantes. La función de los TAD es reagrupar potenciadores y promotores que interactúan juntos dentro de un único dominio funcional grande en lugar de tenerlos diseminados en diferentes TAD.[31] Sin embargo, los estudios del desarrollo del ratón señalan que dos TAD adyacentes pueden regular el mismo grupo de genes. El estudio más relevante sobre la evolución de la extremidad muestra que el TAD en el 5 'del grupo de genes HoxD en los genomas tetrápodos impulsa su expresión en los embriones de yema de la extremidad distal, dando lugar a la mano, mientras que el situado en el lado 3' lo hace en la yema proximal de la extremidad, que da origen al brazo.[32] Aun así, no se sabe si los TAD son una estrategia adaptativa para mejorar las interacciones regulatorias o un efecto de las limitaciones en estas mismas interacciones. Los límites de TAD a menudo están compuestos por genes internos, ARNt, otras secuencias altamente expresadas y Elementos Cortos Intercalados (SINE). Si bien estos genes pueden aprovechar su posición de borde para expresarse de manera ubicua, no están directamente vinculados con la formación de bordes de TAD. Las moléculas específicas identificadas en los límites de los TAD se denominan aislantes o proteínas arquitectónicas porque no solo bloquean la expresión con fugas del potenciador, sino que también garantizan una compartimentación precisa de las entradas reguladoras cis al promotor objetivo. Estos aislantes son proteínas de unión al ADN como CTCF y TFIIIC que ayudan a reclutar socios estructurales como cohesinas y condensinas. La localización y unión de proteínas arquitectónicas a sus correspondientes sitios de unión está regulada por modificaciones postraduccionales. ☃☃ Los motivos de unión al ADN reconocidos por las proteínas arquitectónicas son de alta ocupación y alrededor de una megabase entre sí o de baja ocupación y dentro de los TAD. Los sitios de alta ocupación generalmente se conservan y están estáticos, mientras que los sitios intra-TAD son dinámicos de acuerdo con el estado de la celda, por lo tanto, los TAD mismos están compartimentados en subdominios que pueden denominarse subTAD desde unos pocos kb hasta un TAD largo (19). Cuando los sitios de unión arquitectónicos están a menos de 100 kb entre sí, las proteínas mediadoras son las proteínas arquitectónicas que cooperan con la cohesina. Para subTADs mayores de 100 kb y límites de TAD, CTCF es el aislante típico que interactúa con la cohesión.[33]
Del complejo de preiniciación y escape promotor
En eucariotas, el ARNr ribosómico y los ARNt involucrados en la traducción están controlados por la ARN polimerasa I (Pol I) y la ARN polimerasa III (Pol III). La ARN polimerasa II (Pol II) es responsable de la producción de ARN mensajero (ARNm) dentro de la célula. Particularmente para Pol II, muchos de los puntos de control regulatorios en el proceso de transcripción ocurren en el ensamblaje y escape del complejo de pre-iniciación. Una combinación de factores de transcripción específicos de genes reclutará TFIID y/o TFIIA en el promotor central, seguido de la asociación de TFIIB, creando un complejo estable en el que se pueden ensamblar el resto de los factores de transcripción generales (GTF).[34] Este complejo es relativamente estable y puede sufrir múltiples rondas de iniciación de la transcripción.[35] Después de la unión de TFIIB y TFIID, Pol II, el resto de GTF se pueden ensamblar. Este ensamblaje está marcado por la modificación postraduccional (típicamente fosforilación) del dominio C-terminal (CTD) de Pol II a través de varias quinasas.[36] El CTD es un gran dominio no estructurado que se extiende desde la subunidad RbpI de Pol II y consta de muchas repeticiones de la secuencia de heptada YSPTSPS. TFIIH, la helicasa que permanece asociada con Pol II durante la transcripción, también contiene una subunidad con actividad quinasa que fosforilará las serinas 5 en la secuencia de heptada. De manera similar, tanto CDK8 (una subunidad del complejo mediador multiproteico masivo) como CDK9 (una subunidad del factor de elongación p-TEFb), tienen actividad quinasa hacia otros residuos en el CTD.[37] Estos eventos de fosforilación promueven el proceso de transcripción y sirven como sitios de reclutamiento para la maquinaria de procesamiento de ARNm. Las tres de estas quinasas responden a señales corriente arriba, y la falta de fosforilación de CTD puede conducir a una polimerasa estancada en el promotor.
En cáncer
En los vertebrados, la mayoría de los promotores de genes contienen una isla CpG con numerosos sitios CpG.[38] Cuando muchos de los sitios CpG del promotor de un gen están metilados, el gen se silencia.[39] Los cánceres colorrectales suelen tener de 3 a 6 mutaciones de conductor y de 33 a 66 mutaciones de autoestopista o pasajero.[40] Sin embargo, el silenciamiento transcripcional puede ser más importante que la mutación para provocar la progresión al cáncer. Por ejemplo, en los cánceres colorrectales alrededor de 600 a 800 genes son silenciados transcripcionalmente por la metilación de la isla CpG (regulación de la transcripción en el cáncer). La represión transcripcional en el cáncer también puede ocurrir por otros mecanismos epigenéticos, como la expresión alterada de microARN.[41] En el cáncer de mama, la represión transcripcional de BRCA1 puede ocurrir con mayor frecuencia por microARN-182 sobreexpresado que por hipermetilación del promotor BRCA1 (ver Baja expresión de BRCA1 en cánceres de mama y ovario).
Referencias
- Jacob, F.; Monod, J. (1961-06). «Genetic regulatory mechanisms in the synthesis of proteins». Journal of Molecular Biology 3: 318-356. ISSN 0022-2836. PMID 13718526. doi:10.1016/s0022-2836(61)80072-7.
- Englesberg, E.; Irr, J.; Power, J.; Lee, N. (1965-10). «Positive control of enzyme synthesis by gene C in the L-arabinose system». Journal of Bacteriology 90 (4): 946-957. ISSN 0021-9193. PMID 5321403. doi:10.1128/JB.90.4.946-957.1965.
- Busby, S.; Ebright, R. H. (2 de diciembre de 1994). «Promoter structure, promoter recognition, and transcription activation in prokaryotes». Cell 79 (5): 743-746. ISSN 0092-8674. PMID 8001112. doi:10.1016/0092-8674(94)90063-9.
- Payankaulam, Sandhya; Li, Li M.; Arnosti, David N. (14 de septiembre de 2010). «Transcriptional repression: conserved and evolved features». Current biology: CB 20 (17): R764-771. ISSN 1879-0445. PMC 3033598. PMID 20833321. doi:10.1016/j.cub.2010.06.037.
- Gruber, Tanja M.; Gross, Carol A. (2003). «Multiple sigma subunits and the partitioning of bacterial transcription space». Annual Review of Microbiology 57: 441-466. ISSN 0066-4227. PMID 14527287. doi:10.1146/annurev.micro.57.030502.090913.
- Madigan, Michael T. Brock Biology of Microorganisms, 15e. Pearson. p. 178. ISBN 9780134602295.
- Kang, J.; Mishanina, T. V.; Landick, R.; Darst, S. A. (2019). «Mechanisms of Transcriptional Pausing in Bacteria.». Journal of Molecular Biology. PMID 31310765. doi:10.1016/j.jmb.2019.07.017.
- Zhang, J.; Landick, R. (2016). «A Two-Way Street: Regulatory Interplay between RNA Polymerase and Nascent RNA Structure.». Journal of Molecular Biology. PMC 4911296. PMID 26822487. doi:10.1016/j.tibs.2015.12.009.
- Artsimovitch, I. (2018). «Rebuilding the bridge between transcription and translation.». Molecular Microbiology. PMID 29608805. doi:10.1111/mmi.13964.
- «malE - Maltose-binding periplasmic protein precursor - Escherichia coli (strain K12) - malE gene & protein». www.uniprot.org (en inglés). Consultado el 20 de noviembre de 2017.
- «malF - Maltose transport system permease protein MalF - Escherichia coli (strain K12) - malF gene & protein». www.uniprot.org (en inglés). Consultado el 20 de noviembre de 2017.
- «malG - Maltose transport system permease protein MalG - Escherichia coli (strain K12) - malG gene & protein». www.uniprot.org (en inglés). Consultado el 20 de noviembre de 2017.
- Struhl, K. (9 de julio de 1999). «Fundamentally different logic of gene regulation in eukaryotes and prokaryotes». Cell 98 (1): 1-4. ISSN 0092-8674. PMID 10412974. doi:10.1016/S0092-8674(00)80599-1.
- Calo, Eliezer; Wysocka, Joanna (7 de marzo de 2013). «Modification of enhancer chromatin: what, how, and why?». Molecular Cell 49 (5): 825-837. ISSN 1097-4164. PMC 3857148. PMID 23473601. doi:10.1016/j.molcel.2013.01.038.
- de Napoles, Mariana; Mermoud, Jacqueline E.; Wakao, Rika; Tang, Y. Amy; Endoh, Mitusuhiro; Appanah, Ruth; Nesterova, Tatyana B.; Silva, Jose et al. (2004-11). «Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation». Developmental Cell 7 (5): 663-676. ISSN 1534-5807. PMID 15525528. doi:10.1016/j.devcel.2004.10.005.
- Maeder, Morgan L; Angstman, James F; Richardson, Marcy E; Linder, Samantha J; Cascio, Vincent M; Tsai, Shengdar Q; Ho, Quan H; Sander, Jeffry D et al. (2013-12). «Targeted DNA Demethylation and Endogenous Gene Activation Using Programmable TALE-TET1 Fusions». Nature biotechnology 31 (12): 1137-1142. ISSN 1087-0156. PMC 3858462. PMID 24108092. doi:10.1038/nbt.2726.
- Turek-Plewa, Justyna; Jagodziński, Paweł P. (2005). «The role of mammalian DNA methyltransferases in the regulation of gene expression». Cellular & Molecular Biology Letters 10 (4): 631-647. ISSN 1425-8153. PMID 16341272.
- Lövkvist, Cecilia; Dodd, Ian B.; Sneppen, Kim; Haerter, Jan O. (20 de junio de 2016). «DNA methylation in human epigenomes depends on local topology of CpG sites». Nucleic Acids Research 44 (11): 5123-5132. ISSN 0305-1048. PMC 4914085. PMID 26932361. doi:10.1093/nar/gkw124.
- Jabbari, Kamel; Bernardi, Giorgio (26 de mayo de 2004). «Cytosine methylation and CpG, TpG (CpA) and TpA frequencies». Gene 333: 143-149. ISSN 0378-1119. PMID 15177689. doi:10.1016/j.gene.2004.02.043.
- Weber, Michael; Hellmann, Ines; Stadler, Michael B.; Ramos, Liliana; Pääbo, Svante; Rebhan, Michael; Schübeler, Dirk (2007-04). «Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome». Nature Genetics 39 (4): 457-466. ISSN 1061-4036. PMID 17334365. doi:10.1038/ng1990.
- Yang, Xiaojing; Han, Han; De Carvalho, Daniel D.; Lay, Fides D.; Jones, Peter A.; Liang, Gangning (13 de octubre de 2014). «Gene Body Methylation can alter Gene Expression and is a Therapeutic Target in Cancer». Cancer cell 26 (4): 577-590. ISSN 1535-6108. PMC 4224113. PMID 25263941. doi:10.1016/j.ccr.2014.07.028.
- Vaquerizas, Juan M.; Kummerfeld, Sarah K.; Teichmann, Sarah A.; Luscombe, Nicholas M. (04 2009). «A census of human transcription factors: function, expression and evolution». Nature Reviews. Genetics 10 (4): 252-263. ISSN 1471-0064. PMID 19274049. doi:10.1038/nrg2538.
- Yin, Yimeng; Morgunova, Ekaterina; Jolma, Arttu; Kaasinen, Eevi; Sahu, Biswajyoti; Khund-Sayeed, Syed; Das, Pratyush K.; Kivioja, Teemu et al. (05 05, 2017). «Impact of cytosine methylation on DNA binding specificities of human transcription factors». Science (New York, N.Y.) 356 (6337). ISSN 1095-9203. PMID 28473536. doi:10.1126/science.aaj2239.
- Whiteside, S. T.; Goodbourn, S. (1993-04). «Signal transduction and nuclear targeting: regulation of transcription factor activity by subcellular localisation». Journal of Cell Science. 104 ( Pt 4): 949-955. ISSN 0021-9533. PMID 8314906.
- Vihervaara, Anniina; Sistonen, Lea (15 de enero de 2014). «HSF1 at a glance». Journal of Cell Science 127 (Pt 2): 261-266. ISSN 1477-9137. PMID 24421309. doi:10.1242/jcs.132605.
- Levine, Mike (14 de septiembre de 2010). «Transcriptional enhancers in animal development and evolution». Current biology: CB 20 (17): R754-763. ISSN 1879-0445. PMC 4280268. PMID 20833320. doi:10.1016/j.cub.2010.06.070.
- van Arensbergen, Joris; van Steensel, Bas; Bussemaker, Harmen J. (2014-11). «In search of the determinants of enhancer-promoter interaction specificity». Trends in Cell Biology 24 (11): 695-702. ISSN 1879-3088. PMC 4252644. PMID 25160912. doi:10.1016/j.tcb.2014.07.004.
- Mercer, Tim R.; Mattick, John S. (2013-07). «Understanding the regulatory and transcriptional complexity of the genome through structure». Genome Research 23 (7): 1081-1088. ISSN 1549-5469. PMC 3698501. PMID 23817049. doi:10.1101/gr.156612.113.
- Dekker, Job; Marti-Renom, Marc A.; Mirny, Leonid A. (2013-6). «Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data». Nature reviews. Genetics 14 (6): 390-403. ISSN 1471-0056. PMC 3874835. PMID 23657480. doi:10.1038/nrg3454.
- Gómez-Díaz, Elena; Corces, Victor G. (2014-11). «Architectural proteins: Regulators of 3D genome organization in cell fate». Trends in cell biology 24 (11): 703-711. ISSN 0962-8924. PMC 4254322. PMID 25218583. doi:10.1016/j.tcb.2014.08.003.
- Smallwood, Andrea; Ren, Bing (2013-6). «Genome organization and long-range regulation of gene expression by enhancers». Current opinion in cell biology 25 (3): 387-394. ISSN 0955-0674. PMC 4180870. PMID 23465541. doi:10.1016/j.ceb.2013.02.005.
- Woltering, Joost M.; Noordermeer, Daan; Leleu, Marion; Duboule, Denis (21 de enero de 2014). «Conservation and Divergence of Regulatory Strategies at Hox Loci and the Origin of Tetrapod Digits». PLoS Biology 12 (1). ISSN 1544-9173. PMC 3897358. PMID 24465181. doi:10.1371/journal.pbio.1001773.
- Phillips-Cremins, Jennifer E.; Sauria, Michael E. G.; Sanyal, Amartya; Gerasimova, Tatiana I.; Lajoie, Bryan R.; Bell, Joshua S. K.; Ong, Chin-Tong; Hookway, Tracy A. et al. (6 de junio de 2013). «Architectural protein subclasses shape 3-D organization of genomes during lineage commitment». Cell 153 (6): 1281-1295. ISSN 0092-8674. PMC 3712340. PMID 23706625. doi:10.1016/j.cell.2013.04.053.
- Thomas, Mary C.; Chiang, Cheng-Ming (2006-05). «The general transcription machinery and general cofactors». Critical Reviews in Biochemistry and Molecular Biology 41 (3): 105-178. ISSN 1040-9238. PMID 16858867. doi:10.1080/10409230600648736.
- Voet, Donald Voet, Judith G. (2011). Biochemistry (4th edición). Hoboken, NJ: John Wiley & Sons. ISBN 978-0470917459.
- Napolitano, Giuliana; Lania, Luigi; Majello, Barbara (2014-05). «RNA polymerase II CTD modifications: how many tales from a single tail». Journal of Cellular Physiology 229 (5): 538-544. ISSN 1097-4652. PMID 24122273. doi:10.1002/jcp.24483.
- Chapman, Rob D.; Conrad, Marcus; Eick, Dirk (2005-9). «Role of the Mammalian RNA Polymerase II C-Terminal Domain (CTD) Nonconsensus Repeats in CTD Stability and Cell Proliferation». Molecular and Cellular Biology 25 (17): 7665-7674. ISSN 0270-7306. PMC 1190292. PMID 16107713. doi:10.1128/MCB.25.17.7665-7674.2005.
- Saxonov, Serge; Berg, Paul; Brutlag, Douglas L. (31 de enero de 2006). «A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters». Proceedings of the National Academy of Sciences of the United States of America 103 (5): 1412-1417. ISSN 0027-8424. PMC 1345710. PMID 16432200. doi:10.1073/pnas.0510310103.
- Bird, Adrian (1 de enero de 2002). «DNA methylation patterns and epigenetic memory». Genes & Development 16 (1): 6-21. ISSN 0890-9369. PMID 11782440. doi:10.1101/gad.947102.
- Vogelstein, Bert; Papadopoulos, Nickolas; Velculescu, Victor E.; Zhou, Shibin; Diaz, Luis A.; Kinzler, Kenneth W. (29 de marzo de 2013). «Cancer Genome Landscapes». Science (New York, N.Y.) 339 (6127): 1546-1558. ISSN 0036-8075. PMC 3749880. PMID 23539594. doi:10.1126/science.1235122.
- Tessitore, Alessandra; Cicciarelli, Germana; Del Vecchio, Filippo; Gaggiano, Agata; Verzella, Daniela; Fischietti, Mariafausta; Vecchiotti, Davide; Capece, Daria et al. (2014). «MicroRNAs in the DNA Damage/Repair Network and Cancer». International Journal of Genomics 2014: 820248. ISSN 2314-436X. PMC 3926391. PMID 24616890. doi:10.1155/2014/820248.
Enlaces externos
- Base de datos de factores de transcripción de plantas y plataforma de análisis y datos de regulación de la transcripción de plantas
- MIT: Activando una nueva comprensión de la regulación genética
| Control de autoridades |
|
|---|
 Datos: Q3787274
Datos: Q3787274 Multimedia: Transcriptional regulation / Q3787274
Multimedia: Transcriptional regulation / Q3787274