Sólidos moleculares
Un sólido molecular es un sólido formado por moléculas discretas. Las fuerzas de cohesión que unen las moléculas son las fuerzas de van der Waals, las interacciones dipolo-dipolo, las interacciones cuadrupolo, las interacciones π-π, los enlaces de hidrógeno, los enlaces halógenos, las fuerzas de dispersión de London y, en algunos sólidos moleculares, las interacciones coulombianas.[3][4][5][6][7][8][9][10] Van der Waals, las interacciones dipolares, las interacciones cuadrupolares, las interacciones π-π, los enlaces de hidrógeno y los enlaces halógenos (2-127 kJ mol-1)[10] suelen ser mucho más débiles que las fuerzas que mantienen unidos otros sólidos: metálicos (enlaces metálicos, 400-500 kJ mol-1),[4] iónicos (fuerzas de Coulomb, 700-900 kJ mol-1),[4] y sólidos de red (enlaces covalentes, 150-900 kJ mol-1).[4][10] Las interacciones intermoleculares, por lo general, no implican electrones deslocalizados, a diferencia de los enlaces metálicos y ciertos enlaces covalentes. Las excepciones son los complejos de transferencia de carga, como el tetratiafulvaleno-tetracianoquinodimetano (TTF-TCNQ), una sal iónica radical.[5] Estas diferencias en la fuerza (es decir, covalente frente a van der Waals) y las características electrónicas (es decir, electrones deslocalizados) con respecto a otros tipos de sólidos dan lugar a las propiedades mecánicas, electrónicas y térmicas únicas de los sólidos moleculares.[3][4][5][8]

Los sólidos moleculares son malos conductores eléctricos,[4][5] aunque algunos, como el TTF-TCNQ, son semiconductores (ρ = 5 x 102 Ω-1 cm-1)[5] y, aun así, su conductividad es sustancialmente inferior a la del cobre (ρ = 6 x 105 Ω-1 cm-1).[8] Los sólidos moleculares tienden a tener menor resistencia a la fractura (sacarosa, KIc = 0,08 MPa m1/2)[11] que los metálicos (hierro, KIc = 50 MPa m1/2),[11] iónicos (cloruro sódico, KIc = 0,5 MPa m1/2),[11] y covalentes (diamante, KIc = 5 MPa m1/2).[12] Los sólidos moleculares tienen puntos de fusión (Tm) y ebullición (Tb) bajos en comparación con los sólidos metálicos (hierro), iónicos (cloruro sódico) y covalentes (diamante).[4][5] [8][13] Algunos ejemplos de sólidos moleculares con temperaturas de fusión y ebullición bajas son el argón, el agua, el naftaleno, la nicotina y la cafeína (véase la tabla siguiente).[13][14] Los constituyentes de los sólidos moleculares varían en tamaño desde gases monoatómicos condensados[15] a moléculas pequeñas (por ejemplo, naftaleno y agua)[16][17] a moléculas grandes con decenas de átomos (por ejemplo, fullereno con 60 átomos de carbono).[18]
| Tipo de sólido | Material | Tm (°C) | Tb (°C) |
|---|---|---|---|
| Metálico | Hierro | 1,538[13] | 2,861[13] |
| Iónico | Cloruro de sodio | 801[13] | 1,465[13] |
| Covalente | Diamante | 4,440[13] | - |
| Molecular | Argón | -189.3[13] | -185.9[13] |
| Molecular | Agua | 0[13] | 100[13] |
| Molecular | Naftaleno | 80.1[13] | 217.9[13] |
| Molecular | Nicotina | -79[13] | 491[13] |
| Molecular | Cafeína | 235.6[13] | 519.9[14] |
Composición y estructura
Los sólidos moleculares pueden estar formados por átomos individuales, moléculas diatómicas y/o poliatómicas.[1][2][3][4][5][6][7] Las interacciones intermoleculares entre los constituyentes dictan cómo se estructura la red cristalina del material.[19][20][21] Todos los átomos y moléculas pueden participar en las fuerzas de dispersión de Van der Waals y London (estéricas). Es la ausencia o presencia de otras interacciones intermoleculares en función del átomo o la molécula lo que confiere a los materiales propiedades únicas.[19]
Fuerzas de Van der Waals
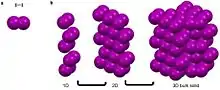
El argón es un gas noble que tiene un octeto completo, no tiene carga y es apolar.[3][4][7][8] Estas características hacen que sea desfavorable para el argón participar en enlaces metálicos, covalentes e iónicos, así como en la mayoría de las interacciones intermoleculares.[3][4][7][8] Aunque puede participar en las fuerzas de dispersión de Van der Waals y London.[3][4] Estas débiles auto-interacciones son isotrópicas y dan lugar a la ordenación de largo alcance de los átomos en un empaquetamiento cúbico centrado en la cara cuando se enfría por debajo de -189,3.[13] Del mismo modo, el yodo, una molécula diatómica lineal tiene un dipolo neto de cero y sólo puede participar en las interacciones de Van der Waals que son bastante isotrópicas.[3][4][7][8] Esto da lugar a la simetría bipirámide.
Interacciones dipolo-dipolo y cuadrupolo

En el caso de la acetona, las interacciones dipolo-dipolo son una de las principales fuerzas impulsoras de la estructura de su red cristalina. El dipolo negativo está causado por el oxígeno. El oxígeno es más electronegativo que el carbono y el hidrógeno,[13] causando una carga parcial negativa (δ-) y positiva (δ+) en el oxígeno y en el resto de la molécula, respectivamente.[3][5] El δ- se orienta hacia el δ+ causando que las moléculas de acetona prefieran alinearse en unas pocas configuraciones en una orientación δ- a δ+ (imagen de la izquierda). El dipolo-dipolo y otras interacciones intermoleculares se alinean para minimizar la energía en el estado sólido y determinan la estructura de la red cristalina.

Un cuadrupolo, como un dipolo, es un polo permanente pero el campo eléctrico de la molécula no es lineal como en la acetona, sino en dos dimensiones.[25] Ejemplos de sólidos moleculares con cuadrupolos son el octafluoronaftaleno y el naftaleno.[17][25] El naftaleno está formado por dos anillos conjugados unidos. La electronegatividad de los átomos de este sistema de anillos y la conjugación provocan una corriente anular que da lugar a un cuadrupolo. En el caso del naftaleno, este cuadrupolo se manifiesta en un δ- y un δ+ que se acumulan dentro y fuera del sistema de anillos, respectivamente.[4][5][6][10][25] El naftaleno se ensambla mediante la coordinación del δ- de una molécula con el δ+ de otra,[4][5][6] lo que da lugar a columnas 1D de naftaleno en un patrón de espiga. A continuación, estas columnas se apilan en capas 2D y luego en materiales a granel 3D. El octafluoronaftaleno sigue este camino de organización para construir materiales a granel, excepto que el δ- y el δ+ están en el exterior y el interior del sistema de anillos, respectivamente.[5]
Enlace de hidrógeno y halógeno
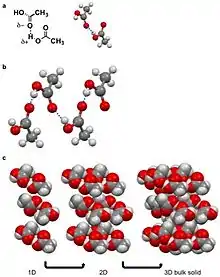
Un enlace de hidrógeno es un dipolo específico en el que un átomo de hidrógeno tiene una carga positiva parcial (δ+) debido a un átomo electronegativo vecino o a un grupo funcional.[9][10] Los enlaces de hidrógeno se encuentran entre las interacciones intermoleculares fuertes que se conocen, aparte de las interacciones ion-dipolo.[10] En los enlaces de hidrógeno intermoleculares, el hidrógeno δ+ interactúa con un δ- en una molécula adyacente. Ejemplos de sólidos moleculares que establecen enlaces de hidrógeno son el agua, los aminoácidos y el ácido acético.[3][5][8][10] En el caso del ácido acético, el hidrógeno (δ+) de la molécula de alcohol del ácido carboxílico establece un enlace de hidrógeno con otra molécula adyacente de la molécula de carbonilo (δ-) del carboxílico. Este enlace de hidrógeno conduce a una cadena de moléculas de ácido acético que se unen por enlace de hidrógeno para minimizar la energía libre.[10][27] Estas cadenas de moléculas de ácido acético se apilan para construir sólidos.
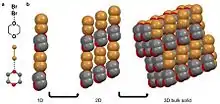
Un enlace halógeno se produce cuando un haluro electronegativo participa en una interacción no covalente con un átomo menos electronegativo de una molécula adyacente.[10][29] Ejemplos de sólidos moleculares que presentan enlaces halógenos son el hexaclorobenceno[11][30] y un cocristal de bromo 1,4-dioxano.[27] En el segundo ejemplo, el átomo de bromo δ- de la molécula de bromo diatómica se alinea con el oxígeno menos electronegativo del 1,4-dioxano. En este caso, el oxígeno se considera δ+ en comparación con el átomo de bromo. Esta coordinación da lugar a una organización en forma de cadena que se apila en 2D y luego en 3D.[27]
Interacciones coulombianas
Las interacciones coulombianas se manifiestan en algunos sólidos moleculares. Un ejemplo bien estudiado es la sal iónica radical TTF-TCNQ con una conductividad de 5 x 102 Ω-1 cm-1,[5] mucho más cercana al cobre (ρ = 6 x 105 Ω-1 cm-1)[8] que muchos sólidos moleculares.[32][18][33] La interacción coulombiana en el TTF-TCNQ se debe a la gran carga negativa parcial (δ = -0,59) de la molécula ciano del TCNQ a temperatura ambiente.[5] Como referencia, una molécula completamente cargada δ = ±1.[5] Esta carga negativa parcial provoca una fuerte interacción con la molécula tiol del TTF. La fuerte interacción conduce a una alineación favorable de estos grupos funcionales adyacentes entre sí en estado sólido,[5][32] mientras que las interacciones π-π hacen que el TTF y el TCNQ se apilen en columnas separadas.[10][32]
Alótropos
Una forma de un elemento puede ser un sólido molecular, pero otra forma de ese mismo elemento puede no ser un sólido molecular.[3][4][5] Por ejemplo, el fósforo sólido puede cristalizar como diferentes alótropos llamados fósforo "blanco", "rojo" y "negro". El fósforo blanco forma cristales moleculares compuestos por moléculas tetraédricas P4.[34] El calentamiento a presión ambiente hasta 250 °C o la exposición a la luz solar convierte el fósforo blanco en fósforo rojo, donde los tetraedros P4 ya no están aislados, sino conectados por enlaces covalentes en cadenas similares a polímeros.[35] El calentamiento del fósforo blanco a altas presiones (GPa) lo convierte en fósforo negro, que tiene una estructura en capas similar al grafito.[36][37] Las transiciones estructurales en el fósforo son reversibles: al liberar alta presión, el fósforo negro se convierte gradualmente en el fósforo rojo, y vaporizando el fósforo rojo a 490 °C en una atmósfera inerte y condensando el vapor, el fósforo rojo covalente puede transformarse en el sólido molecular, el fósforo blanco.[38]
 |
 |
 | ||
|---|---|---|---|---|
| Muestras de fósforo blanco, rojo, violeta y negro | Unidad de estructura de fósforo blanco | Estructuras de color rojo | Violeta | y fósforo negro |
Del mismo modo, el arsénico amarillo es un sólido molecular compuesto por unidades As4.[39] Algunas formas de azufre y selenio están compuestas por unidades S8 (o Se8) y son sólidos moleculares en condiciones ambientales, pero se convierten en alótropos covalentes que tienen cadenas atómicas que se extienden por todo el cristal.[40][41]
Propiedades
Dado que los sólidos moleculares se mantienen unidos por fuerzas relativamente débiles, tienden a tener puntos de fusión y ebullición bajos, baja resistencia mecánica, baja conductividad eléctrica y escasa conductividad térmica.[3][4][5][6][7][8] Además, dependiendo de la estructura de la molécula, las fuerzas intermoleculares pueden tener direccionalidad, lo que conduce a la anisotropía de ciertas propiedades.[4][5][8]
Puntos de fusión y ebullición
El punto de fusión característico de los metales y los sólidos iónicos es de ~ 1000 °C y superior, mientras que los sólidos moleculares suelen fundirse cerca de los 300 °C (véase la tabla), por lo que muchas sustancias correspondientes son líquidas (hielo) o gaseosas (oxígeno) a temperatura ambiente[4][6][7][8][42] Esto se debe a los elementos implicados, las moléculas que forman y las débiles interacciones intermoleculares de las moléculas.
Puntos de fusión de algunos sólidos moleculares[43][44]
- Véase también alcanos superiores
Los alótropos del fósforo son útiles para seguir demostrando esta relación estructura-propiedades. El fósforo blanco, un sólido molecular, tiene una densidad relativamente baja de 1,82 g/cm3 y un punto de fusión de 44,1 °C; es un material blando que se puede cortar con un cuchillo. Cuando se convierte en el covalente fósforo rojo, la densidad pasa a 2,2-2,4 g/cm3 y el punto de fusión a 590 °C y, cuando el fósforo blanco se transforma en el (también covalente) fósforo negro, la densidad pasa a ser de 2,69-3,8 g/cm3 y, la temperatura de fusión, ~200 °C. Tanto la forma de fósforo rojo como la de fósforo negro son significativamente más duras que el fósforo blanco.[45]
Propiedades mecánicas
Los sólidos moleculares pueden ser dúctiles o quebradizos, o una combinación de ambos, dependiendo de la cara del cristal sometida a tensión.[5][11] Tanto los sólidos dúctiles como los quebradizos experimentan una deformación elástica hasta que alcanzan el límite elástico.[8][11] Una vez alcanzado el límite elástico, los sólidos dúctiles experimentan un periodo de deformación plástica y, finalmente, se fracturan.[8][11] Los sólidos frágiles se fracturan inmediatamente después de superar el límite elástico.[8][11] Debido a la estructura asimétrica de la mayoría de las moléculas, muchos sólidos moleculares tienen fuerzas intermoleculares direccionales.[11] Este fenómeno puede dar lugar a propiedades mecánicas anisótropas. Normalmente, un sólido molecular es dúctil cuando tiene interacciones intermoleculares direccionales. Esto permite la dislocación entre las capas del cristal de forma muy parecida a los metales.[5][8][11]
Un ejemplo de sólido molecular dúctil, que puede doblarse 180°, es el hexaclorobenceno (HCB).[11][30] En este ejemplo, las interacciones π-π entre los núcleos de benceno son más fuertes que las interacciones halógenas de los cloruros. Esta diferencia conduce a su flexibilidad.[11][30] Esta flexibilidad es anisotrópica; para doblar el HCB a 180° hay que tensionar la cara [001] del cristal.[30] Otro ejemplo de sólido molecular flexible es el ácido 2-(metiltio)nicotínico (MTN).[11][30] El MTN es flexible debido a su fuerte enlace de hidrógeno y a las interacciones π-π que crean un conjunto rígido de dímeros que se dislocan a lo largo de la alineación de sus metilos terminales.[30] Cuando se tensiona en la cara [010] este cristal se dobla 180°.[30] Nótese que no todos los sólidos moleculares dúctiles se doblan 180° y algunos pueden tener más de una cara de flexión.[30]
Propiedades eléctricas
En general, los sólidos moleculares son aislantes.[5][18] Esta gran brecha de banda (comparada con la del germanio de 0,7 eV)[8] se debe a las débiles interacciones intermoleculares, que dan lugar a una baja movilidad de los portadores de carga. Algunos sólidos moleculares presentan conductividad eléctrica, como TTF-TCNQ con ρ = 5 x 102 Ω-1 cm-1, pero en estos casos el solapamiento orbital es evidente en la estructura cristalina. Los fullerenos, que son aislantes, se vuelven conductores o incluso superconductores al doparlos.[46]
Propiedades térmicas
Los sólidos moleculares tienen muchas propiedades térmicas: capacidad calorífica específica, expansión térmica y conductancia térmica, por nombrar algunas.[3][5][6][7][8] Estas propiedades térmicas vienen determinadas por las vibraciones intra e intermoleculares de los átomos y moléculas del sólido molecular. Aunque las transiciones de un electrón contribuyen a las propiedades térmicas, su contribución es insignificante en comparación con la contribución vibracional.[5][8]
Véase también
Referencias
- Simon, A.; Peters, K. (15 de noviembre de 1980). «Single-crystal refinement of the structure of carbon dioxide». Acta Crystallographica Section B: Structural Crystallography and Crystal Chemistry (en inglés) 36 (11): 2750-2751. ISSN 0567-7408. doi:10.1107/S0567740880009879. Consultado el 8 de agosto de 2023.
- Lehmann, C. W.; Stowasser, Frank (2007). «The Crystal Structure of Anhydrous Beta-Caffeine as Determined from X-ray Powder-Diffraction Data». Chemistry: A European Journal (en inglés): 2908-2911. PMID 17200930. doi:10.1002/chem.200600973.
- Hall, George (1965). «Molecular Solid State Physics». Springer-Verlag.
- Fahlman, B. D. (2011). «Materials Chemistry». Springer.
- Schwoerer, M.; Wolf, H. C. (2007). «Organic Molecular Solids». Wiley-VCH.
- Omar, M. A. (2002). «Elementary Solid State Physics». Londres, Inglaterra: Pearson.
- Patterson, J.; Bailey, B. (2010). «Solid-State Physics». Berlín, Alemania: Springer.
- Turton, R. (2010). «The Physics of Solids». New York, New York: Oxford University Press Inc.
- Keer, H. V. (1993). «Principles of Solid State». Hoboken, New Jersey: Wiley Eastern Limited.
- Israelachvili, J. N. (2011). «Intermolecular and Surface Forces». Cambridge, Massachusetts: Academic Press.
- Varughese, S.; Kiran, M. S. R. N.; Ramamurty, U.; Desiraju, G. R. (2013). «Nanoindentation in Crystal Engineering: Quantifying Mechanical Properties of Molecular Crystals». Angewandte Chemie International Edition: 2701-2712. PMID 23315913. doi:10.1002/anie.201205002.
- Field, J. E., ed. (1979). «The Properties of Diamonds». New York, New York: Academic Press.
- Haynes, W. M.; Lise, D. R.; Bruno, T. J., eds. (2016). «CRC Handbook of Chemistry and Physics». Boca Raton, Florida: CRC Press.
- O'Neil, M. J., ed. (2013). «The Merck Index - An Encyclopedia of Chemicals, Drugs, and Biologicals». Reino Unido: Royal Society of Chemistry.
- Barret, C. S.; Meyer, L. (1965). Daunt, J. G. (ed.). (1965). «Low Temperature Physics: The Crystal Structures of Argon and Its Alloys.». New York, New York: Springer.
- Eisenberg, D.; Kauzmann, W. (2005). «The Structures and Properties of Water». Oxford, UK: Oxford University Press.
- Harvey, G. R. (1991). «Polycyclic Aromatic Hydrocarbons: Chemistry and Carcinogenicity». UK: Cambridge University Press.
- Jones, W., ed. (1997). «Organic Molecular Solids: Properties and Applications». Boca Raton: CRC Press.
- Desiraju, G. R. (2013). «Crystal Engineering: From Molecular to Crystal». Journal of the American Chemical Society: 9952-9967. PMID 23750552. doi:10.1021/ja403264c.
- Thakur, T. S.; Dubey, R.; Desiraju, G. R. (2015). «Crystal Structure and Prediction». Annual Review of Physical Chemistry: 21-42. PMID 25422850. doi:10.1146/annurev-physchem-040214-121452.
- Davey, R. J.; Schroeder, S. L.; Horst, J. H. T. (2013). «Nucleation of Organic Crystals - A Molecular Perspective». Angewandte Chemie International Edition: 2166-2179. PMID 23307268. doi:10.1002/anie.201204824.
- Harris, Harris; Edward, M.; Blake, F. C. (1928). «The Atomic Arrangement of Orthorhombic Iodine». Journal of the American Chemical Society: 1583-1600. doi:10.1021/ja01393a009.
- Allan, D. R.; Clark, S. J.; Ibberson, R. M.; Parsons, S.; Pulham, C. R.; Sawyer, L. «The Influence of Pressure and Temperature on the Crystal Structure of Acetone». semanticscholar.org. Chemical Communications. pp. 751-752. doi:10.1039/A900558G. Consultado el 8 de agosto de 2023.
- Alt, H. C.; Kalus, J. (15 de octubre de 1982). «X-ray powder diffraction investigation of naphthalene up to 0.5 GPa». Acta Crystallographica Section B: Structural Crystallography and Crystal Chemistry (en inglés) 38 (10): 2595-2600. ISSN 0567-7408. doi:10.1107/S056774088200942X. Consultado el 8 de agosto de 2023.
- Williams, J. H. (1993). «The Molecular Electric QuadrupoleMoment and Solid-State Architecture». Accounts of Chemical Research: 593-598. doi:10.1021/ar00035a005.
- Dawson, A.; Allan, D. R.; Parsons, S.; Ruf, M. (1 de junio de 2004). «Use of a CCD diffractometer in crystal structure determinations at high pressure». Journal of Applied Crystallography (en inglés) 37 (3): 410-416. ISSN 0021-8898. doi:10.1107/S0021889804007149. Consultado el 9 de agosto de 2023.
- Dawson, A.; Allan, D. R.; Parsons, S.; Ruf, M. (1 de junio de 2004). «Use of a CCD diffractometer in crystal structure determinations at high pressure». Journal of Applied Crystallography (en inglés) 37 (3): 410-416. ISSN 0021-8898. doi:10.1107/S0021889804007149. Consultado el 9 de agosto de 2023.
- Hassel, O.; Hvoslef, J.; Vihovde, E. Hadler; Sörensen, Nils Andreas (1954). «The Structure of Bromine 1,4-Dioxanate.». Acta Chemica Scandinavica (en inglés) 8: 873-873. ISSN 0904-213X. doi:10.3891/acta.chem.scand.08-0873. Consultado el 9 de agosto de 2023.
- Metrangolo, P.; Meyer, F.; Pilati, Tullio; Resnati, G.; Terraneo, G. (2008). «Halogen Bonding in Supramolecular Chemistry». Angewandte Chemie International Edition. PMID 18651626. doi:10.1002/anie.200800128.
- Reddy, C. M.; Krishan, G. R.; Ghosh, S. (2010). «Mechanical properties of molecular crystals—applications to crystal engineering». CrystEngComm: 2296-2314. doi:10.1039/c003466e.
- Kistenmacher, T. J.; Phillips, T. E.; Cowan, D. O. (15 de marzo de 1974). «The crystal structure of the 1:1 radical cation–radical anion salt of 2,2'-bis-l,3-dithiole (TTF) and 7,7,8,8-tetracyanoquinodimethane (TCNQ)». Acta Crystallographica Section B: Structural Crystallography and Crystal Chemistry (en inglés) 30 (3): 763-768. ISSN 0567-7408. doi:10.1107/S0567740874003669. Consultado el 9 de agosto de 2023.
- Cohen, M. J.; Coleman, L. B.; Garito, A. F.; Heeger, A. J. (1974). «Electrical Conductivity of Tetrathiofulvalinium Tetracyanoquinodimethane (TTF) (TCNQ)». Physical Review B: 1298-1307. doi:10.1103/PhysRevB.10.1298.
- Kistenmacher, T. J.; Phillips, T. E.; Cowan, D. O. (15 de marzo de 1974). «The crystal structure of the 1:1 radical cation–radical anion salt of 2,2'-bis-l,3-dithiole (TTF) and 7,7,8,8-tetracyanoquinodimethane (TCNQ)». Acta Crystallographica Section B: Structural Crystallography and Crystal Chemistry (en inglés) 30 (3): 763-768. ISSN 0567-7408. doi:10.1107/S0567740874003669. Consultado el 9 de agosto de 2023.
- Olmsted, John; Williams, Gregory M. (1997). Chemistry: The Molecular Science (en inglés). Jones & Bartlett Learning. ISBN 978-0-8151-8450-8. Consultado el 9 de agosto de 2023.
- Atul, Singhal (2009). The Pearson Guide to Objective Chemistry for the AIEEE (en inglés). Pearson Education India. ISBN 978-81-317-1359-4. Consultado el 9 de agosto de 2023.
- Wulfsberg, Gary (29 de mayo de 1991). Principles Of Descriptive Inorganic Chemistry (en inglés). University Science Books. ISBN 978-0-935702-66-8. Consultado el 9 de agosto de 2023.
- Simon, Arndt; Borrmann, Horst; Horakh, Jörg (1997). «On the Polymorphism of White Phosphorus». Chemische Berichte: 1235. doi:10.1002/cber.19971300911.
- AK Srivastava and PC Jain. «Chemistry Vol (1 and 2)». FK Publications: 548. ISBN 978-81-88597-83-3.
- Holleman, Arnold F; Wiberg, Egon; Wiberg, Nils (1985). «Arsen». Lehrbuch der Anorganischen Chemie (en alemán): 675-681. ISBN 978-3-11-007511-3.
- «Allotropes - Chemistry Encyclopedia - structure, reaction, elements, metal, gas, number, name, molecule». www.chemistryexplained.com. Consultado el 9 de agosto de 2023.
- James E. House (2008). «Inorganic chemistry». Academic Press: 524. ISBN 978-0-12-356786-4.
- Ebbing, Darrell; Gammon, Steven D. (3 de diciembre de 2007). General Chemistry (en inglés). Cengage Learning. ISBN 978-0-618-85748-7. Consultado el 9 de agosto de 2023.
- Wei, James (4 de enero de 2007). Product Engineering: Molecular Structure and Properties (en inglés). Oxford University Press. ISBN 978-0-19-534792-0. Consultado el 9 de agosto de 2023.
- Lide, D. R., ed. (2005). «CRC Handbook of Chemistry and Physics». Boca Raton (FL): CRC Press. ISBN 0-8493-0486-5.
- AK Srivastava and PC Jain. «Chemistry Vol (1 and 2)». FK Publications: 550. ISBN 978-81-88597-83-3.
- O. Gunnarsson (1997). «Superconductivity in Fullerides». Reviews of Modern Physics (en inglés): 575. doi:10.1103/RevModPhys.69.575.