Señalización paracrina
La señalización paracrina es una forma de señalización celular en la que una célula secreta una molécula de señalización que induce cambios en las células cercanas, alterando el comportamiento o la diferenciación celular de esas células. Las moléculas conocidas como factores paracrinos se difunden sobre distancias relativamente cortas y ejercen su acción sobre células vecinas, a diferencia de los factores endocrinos, que viajan distancias considerables por el sistema circulatorio), las interacciones juxtacrinas, y la señalización autocrina.
El efecto de los factores paracrinos en las células circundantes depende del gradiente de su concentración. La distancia exacta que puede viajar un factor paracrino no se conoce con certeza. Entre los ejemplos de moléculas paracrinas se encuentran las citokinas, el factor de la coagulación espermática, el factor de crecimiento, la somatostatina y la histamina, entre otros.
Para que los factores paracrinos puedan inducir una respuesta en la célula receptora, esta debe poseer los receptores adecuados en la membrana celular. A pesar del rango diverso de respuestas que presentan las células inducidas tras la unión del factor paracrino a su receptor, la mayor parte de los factores paracrinos utilizan un grupo relativamente pequeño y conservado de receptores y rutas de señalización, incluso en diferentes órganos y especies. Las vías y receptores principales pueden ser organizados en cuatro familias principales de acuerdo a la similitud de sus estructuras: la familia del factor de crecimiento de fibroblastos (FGF), la familia Hedgehog, la familia Wnt, y la superfamilia TGF-β.
Familias de receptores
Factor de crecimiento de fibroblastos (FGF)
Aunque la familia de factores paracrinos FGF tienen un amplio rango de funciones, los mayores descubrimientos soportan la idea de que principalmente estimulan la proliferación y diferenciación celular.[1][2] Para desempeñar tan diversas funciones, los FGF pueden ser ayustados en forma alternativa, o incluso tener diferentes codones de iniciación, consiguiendo de esta forma crear cientos de diferentes isoformas de FGF.[3]
Una de las funciones más importantes de los receptores para FGF (FGFR) se encuentra en el desarrollo de las extremidades. Esta señalización involucra nueve isoformas diferentes del receptor obtenidos por ayuste diferencial.[4] Los Fgf8 y Fgf10 son dos jugadores críticos en el desarrollo de las extremidades. En la iniciación del desarrollo de las extremidades y en el crecimiento de las mismas en ratones, las señales axiales provenientes del mesodermo intermedio producen Tbx5, el cual subsecuentemente envía una señal al mesodermo para producir Fgf10. El Fgf10 posteriormente señaliza al ectodermo que comience la producción de Fgf8, el cual además estimula la producción de Fgf10. La deleción de Fgf10 provoca ratones sin miembros.[5]
Además, la señalización paracrina de Fgf es esencial para el desarrollo del ojo de los pollos. El ARNm del fgf8 aparece localizado en donde se diferencia la retina neural de la copa óptica. Estas células se encuentran en contacto con las células del ectodermo externo, las cuales finalmente se convierten en el cristalino.[3]
Fenotipo y supervivencia de los ratones luego del nocaut de algunos genes FGFR:[4]
| Gen FGFR nocaut | Supervivencia | Fenotipo |
|---|---|---|
| Fgf1 | Viable | Poco claro |
| Fgf3 | Viable | Oído interno, diferenciación esquelética (cola) |
| Fgf4 | Letal | Proliferación de la masa interna de células |
| Fgf8 | Letal | Defecto en la gastrulación, desarrollo del SNC, desarrollo de extremidades |
| Fgf10 | Letal | Desarrollo de múltiples órganos (incluyendo miembros, timo, pituitaria) |
| Fgf17 | Viable | Desarrollo cerebelar |
Vía de los Receptores Tirosina Quinasa (RTK)
La señalización paracrina a través de los factores de crecimiento de fibroblastos (FGF) y sus respectivos receptores (FGFR) hacen uso de la vía de receptores tirosina quinasa. Esta vía de señalización ha sido muy estudiada, utilizando los ojos de Drosophila y los cánceres humanos.[6]
La unión de los FGF a los FGFR fosforila a la quinasa inactiva y activa la vía de las RTK. Esta vía comienza en la superficie de la membrana celular, donde un ligando se une a su receptor específico. Los ligandos que se unen a los RTKs incluyen los factores de crecimiento de fibroblastos, factores de crecimiento epidérmico, factores de crecimiento derivados de plaquetas, y factor de células madres.[6] Esto provoca que el receptor transmembrana se dimerice con otro receptor RTK, lo que causa la autofosforilación y el subsecuente cambio conformacional del receptor homodimerizado. Este cambio conformacional activa el sitio quinasa inactivo de cada RTK sobre un residuo de tirosina. Debido al hecho de que el receptor se extiende atravesando la membrana a partir del ambiente extracelular, a través de la bicapa lipídica, y al interior del citoplasma, la unión del receptor al ligando además causa la transfosforilación del dominio citoplasmático del receptor.[7]
Una proteína adaptadora (tal como la SOS) reconoce a la tirosina fosforilada en el receptor. Esta proteína funge como puente que conecta al RTK a una proteína intermedia (tal como la GNRP), comenzando la cascada de señalización intracelular. Mientras tanto, la proteína intermedia estimula a la Ras unida a GDP para formar Ras activa unida a GTP. La GAP finalmente regresa a la Ras a su estado inactivo. La activación de la Ras tiene el potencial de iniciar tres vías de señalización corriente abajo: la vía Ras→Raf→MAP quinasa, la vía de la quinasa PI3, y la vía de la Ral. Cada vía conduce a la activación de factores de transcripción los cuales ingresan al núcleo para alterar la expresión de genes.[8]
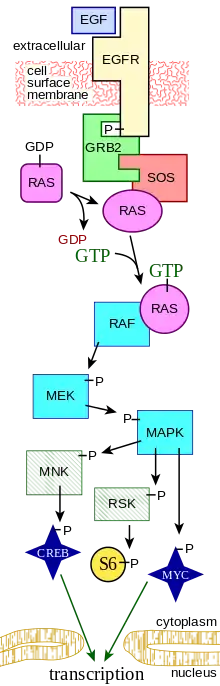
- El receptor RTK y el cáncer
Se ha demostrado que la señalización paracrina de factores de crecimiento entre células vecinas exacerba la carcinogénesis. De hecho las formas mutantes de RTK desemparejados pueden desempeñar un rol causal en varios tipos diferentes de cáncer. Los protooncogenes Kit codifican para un receptor de tipo tirosina quinasa cuyo ligando es una proteína paracrina llamada factor de célula stem (SCF), el cual es importante en la hematopoyesis (la génesis de las células de la sangre).[9] El receptor Kit y los receptores tipo tirosina quinasa relacionados de hecho son inhibitorios y suprimen efectivamente el disparo del receptor. Las formas mutantes del recepto Kit, las cuales se disparan constitutivamente en forma independiente a la unión con su ligando, se han encontrado en varios tipos de neoplasias malignas.[10]
- Vía del RTK y cáncer
Las investigaciones sobre cáncer de tiroides han contribuido a la noción de que la señalización paracrina podría ayudar a crear un microambiente dentro de los tumores. La transcripción de quimiocinas se encuentra regulada al alza cuando la proteína Ras se encuentra en su estado basal unida a GTP. Las quimiocinas se liberan entonces de las células, libres para unirse a otra célula cercana. La señalización parácrina entre células vecinas crea este bucle de retroalimentación positiva. De esta forma, la transcripción constitutiva de proteínas reguladas al alza forma un ambiente ideal para el desarrollo de los tumores.[11] Efectivamente, la unión de múltiples ligandos al receptor RTK sobreestimula la vía Ras-Raf-MAPK, la cual sobreexpresa la capacidad mitogénica e invasiva de las células.[12]
Vía Jak-STAT
Además de a la vía RTK, los factores de crecimiento de fibroblastos pueden activar también a la cascada de señalización Jak-STAT. En lugar de contener dominios tirosina quinasa asociados covalentemente, los receptores Jak-STAT forman complejos no covalentes con las tirosina quinasas de la clase Jak (Janus quinasa). Esta clase de receptores se unen por ejemplo a la eritropoyetina (importante para la eritropoyesis), trombopoyetina (importante para la formación de plaquetas, e interferón (importante para la función inmune mediada por células).[13]
Luego de la dimerización de los receptores de citoquinas que sigue a la unión a sus ligandos, los receptores Jak se transfosforilan uno a otro. Las fosfotirosinas resultantes atraen a proteínas STAT. Las proteínas STAT se dimerizan e ingresan al núcleo para actuar como factores de transcripción alterando la expresión de genes.[13] En particular las proteínas STAT transcriben genes que contribuyen a la proliferación y supervivencia celular, tales como los genes myc.[14]
Fenotipo y supervivencia de ratones luego del noqueo de algunos genes Jak o STAT:[15]
| Gen noqueado | Supervivencia | Fenotipo |
|---|---|---|
| Jak1 | Letal | Déficits neurológicos |
| Jak2 | Letal | Fallas en la eritropoyesis |
| Stat1 | Viable | Enanismo humano y síndromes de craneosinostosis |
| Stat3 | Letal | Fenotipos de tejidos específicos |
| Stat4 | Viable | Diferenciación defectuosa de Th1 dependiente de IL-12, aumento en la susceptibilidad a patógenos intracelulares |
- Vías Jak-STAT aberrantes y mutaciones óseas
La vía Jak-STAT es un instrumento fundamental en el desarrollo de las extremidades, específicamente en su capacidad para regular el crecimiento óseo a través de la señalización parácrina por medio de citoquinas. Sin embargo las mutaciones en esta vía, han sido implicadas en varias formas de enanismo: enanismo acondroplásico (viable) y displasia tanatofórica (letal). Esta última es debido a una mutación en el gen del factor de crecimiento de fibroblastos, que causa una activación prematura y constitutiva del factor de transcripción Stat1. Como resultado la división de los condrocitos termina prematuramente, resultando en un enanismo letal. Las células de las placas de crecimiento de costillas y miembros no lo transcriben. Como resultado, la incapacidad de la caja torácica para expandirse evita que el feto pueda acomodar los órganos del tórax y en caso de llegar al nacimiento evita que pueda respirar.[17]
- La vía Jak-STAT y el cáncer
Las investigaciones es las vías de señallización Jak-STAT revelaron su potencial en la activación de comportamiento invasivo de las células epiteliales ováricas. Esta transición de epitelial a mesenquimal es altamente evidente en las metástasis.[18] La señalización paracrina a través de la vía Jak-STAT es necesaria para la transición de células epiteliales estáticas a células mesenquimatosas móviles, las cuales son capaces de invadir los tejidos aledaños. Hasta el momento se ha encontrado que solo la vía Jak-STAT es capaz de inducir células migratorias.[19]
Familia Hedgehog
La familia de proteínas hedgehog se encuentra involucrada en la inducción de los tipos celulares y en la creación de límites de tejidos y patrones; y se encuentran en todos los organismos bilaterales. Las proteínas hedgehog fueron descubiertas por primera vez y estudiadas en Drosophila. Las proteínas hedgehog producen señales clave para el establecimiento y desarrollo de los miembros, el plan general de construcción del organismo de las mosquitas de la fruta; como así también en la homeostasis de los tejidos adultos, se encuentran involucradas en la embriogénesis tardía y en la metamorfosis. Se han encontrado al menos tres homólogos de las proteínas hedgehog en los vertebrados: la sonic hedgehog, desert hedgehog e indian hedgehog. La proteína sonic hedgehog (SHH) posee varios roles en el desarrollo de los vertebrados, donde media la señalización y regulación de la organización del sistema nervioso central, extremidades y la polaridad en la embriogénesis de los somites. La proteína desert hedgehog (DHH) se expresa en las células de Sertoli, las que participan en la espermatogénesis. Finalmente la proteína indian hedgehog (IHH) se expresa en intestino y cartílago, donde desempeña un importante papel en el crecimiento óseo posnatal.[20][21][22]
Vía de señalización hedgehog

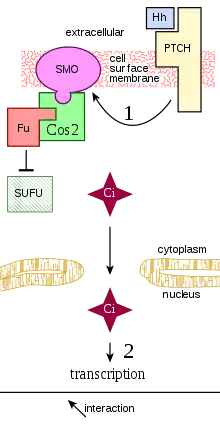
Los miembros de la familia de proteínas Hedgehog actúan por medio de la unión a un receptor "Patched" transmembrana, el cual se encuentra unido a la proteína "Smoothened", por medio de la cual la señal Hedgehog puede ser transducida. En ausencia de Hedgehog, el receptor Patched inhibe la acción de Smoothened. La inhibición de Smoothened produce que el complejo formado por las proteínas Cubitus interruptus (Ci), Fused, y Cos que se encuentra unido a los microtúbulos permanezca intacto. En esta conformación, la proteína Ci se escinde de forma tal que una porción de la proteína se capacita para entrar al núcleo y actuar como un represor transcripcional. En presencia de Hedgehog, Patched ya no es capaz de inhibir a Smoothened. Luego la proteína Smoothened activa es capaz de inhibir a la PKA y Slimb, de forma tal que la proteína Ci no resulta escindida. Esta proteína Ci intacta puede ingresar al núcleo, asociada con la proteína CPB y actuar como activador transcripcional, induciendo la expresión de los genes que responden a Hedgehog.[22][23][24]
- Vía de señalización Hedgehog y cáncer
La vía de señalización Hedgehog es crítica para un apropiado bosquejado y orientación de los tejidos durante el desarrollo normal de la mayor parte de los animales. Las proteínas Hedgehog inducen la proliferación celular de ciertos tipos celulares y la diferenciación de otros. La activación aberrante de la vía Hedgehog ha sido implicada en varios tipos de cáncer, en particular de los carcinomas de células basales. Esta activación descontrolada de las proteínas Hedgehog puede ser causada por mutaciones en la vía de la señal, la cual puede ser independiente de ligando, o una mutación que causa la sobreexpresión de la proteína Hedgehog, la cual es dependiente del ligando. Esta conexión entre la vía de señalización Hedgehog y los cánceres humanos pueden resultar providenciales para una posible intervención terapéutica como así para el tratamiento de estos tipos de cáncer. La vía de señalización Hedgehog se encuentra involucrada en la regulación normal de las poblaciones de células madre, y es necesaria para la regeneración y crecimiento de los órganos dañados. Esta puede ser otra posible ruta para la tumorogénesis involucrando a la vía Hedgehog.[25][26][27]
Familia Wnt
La familia de proteínas Wnt incluye un gran número de glicoproteínas ricas en cisteína. Las proteínas Wnt activan cascadas de transducción de señales por medio de tres diferentes vías, la vía Wnt canónica, la vía Wnt no canónica de polaridad celular planar, y la vía no canónica Wnt/Ca2+
. Las proteínas Wnt parecen controlar un amplio rango de procesos de desarrollo y han mostrado ser necesarias para el control de la orientación del huso acromático, polaridad celular, adhesión mediada por caderinas, y desarrollo temprano de embriones de muchos organismos diferentes. Las investigaciones actuales han indicado que la desrregulación de la señalización Wnt desempeña un rol en la formación de tumores, porque a nivel celular, las proteínas Wnt a menudo regulan la proliferación celular, morfología celular, motilidad celular, y destino celular.[28]
Vía de señalización Wnt canónica
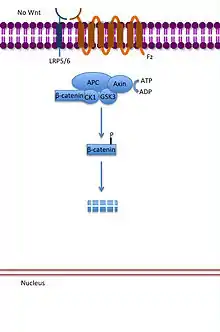
En la vía canónica, las proteínas Wnt se unen a su receptor transmembrana de la familia de proteínas Frizzled. La unión de Wnt a las proteínas Frizzled activa a la proteína Dishevelled. En su estado activo al proteína Dishevelled inhibe la actividad de la enzima glicógeno sintasa quinasa 3 (GSK3). Normalmente la GSK3 activa previene la disociación de la β-catenina de la proteína APC, lo que desembocó en la degradación de la β-catenina. De esta forma la GSK3 inhibida, permite a la β-catenina disociarse de la APC, acumularse, y migrar hacia el núcleo. En el núcleo la β-catenina se asocia al factor de transcripción Lef/Tcf, el cual se encuentra funcionando como un represor del ADN, inhibiendo al transcripción de los genes a los que se encuentra unido. La unión de la β-catenina al Lef/Tcf funciona como un activador de la transcripción, activando la transcripción de los genes que responden a Wnt.[29][30][31]
Vías de señalización Wnt no canónicas
Las vías de señalización Wnt no canónicas proveen una vía de transducción para Wnt que no involucra a la β-catenina. En las vías no canónicas, Wnt afecta al citoesqueleto de actina y microtúbulos como así también a la transcripción génica.
- La vía no canónica de polaridad celular planar (PCP)

La vía no canónica PCP regula la morfología celular, la división celular y la motilidad celular. En esta vía, nuevamente, las proteínas Wnt se unen a Frizzled y la activan, de tal forma que Frizzled activa a la proteína Dishevelled que se encuentra anclada a la membrana celular por medio de una proteína Prickle y una proteína Stbm transmembrana. La proteína Dishelvelled activada a su vez activa la proteína RhoA (una GTPasa) a través del activador de morfogénesis 1 asociado a Dishelvelled (Daam1) y la proteína Rac. La proteína RohA activada se encuentra de esta forma capacitada para inducir cambios en el citoesqueleto activando a la quinasa asociada a RohA (ROCK), y de esta forma afectar directamente a la transcripción de genes. La Rac activada puede inducir directamente cambios en el citoesqueleto y afectar a la transcripción de genes a través de la activación de JNK.[29][30][31]
- La vía no canónica Wnt/Ca2+
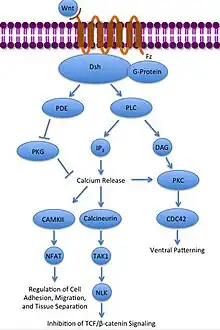
La vía no canónica Wnt/Ca2+
regula los niveles intracelulares de calcio. En esta vía nuevamente Wnt se una a Frizzled y la activa. Sin embargo, en este caso la proteína Frizzled activada provoca que una proteína G acoplada active a una fosfolipasa (PLC), la cual interactúa con PIP
2 y lo escinde en DAG e IP
3. El IP
3 puede luego unirse a un receptor en el retículo endoplasmático para inducir la liberación intracelular de los depósitos de calcio, para de esta forma inducir la expresión de genes dependiente de calcio.[29][30][31]
Vía de señalización Wnt y cáncer
Las vías de señalización Wnt son críticas en la comunicación y señalización célula-célula durante la embriogénesis y el desarrollo normal y es necesario para el mantenimiento del tejido adulto; por lo tanto no es difícil entender por qué una disrupción en las vías de señalización Wnt puede promover la aparición de enfermedades degenerativas y cáncer.
Las vías de señalización Wnt son complejas, implican a varios elementos diferentes y por lo tanto poseen numerosas dianas donde pueden presentarse defectos en la regulación. Las mutaciones que provocan una activación constitutiva de las vías Wnt pueden desembocar en la formación de tumores y cáncer. La activación aberrante de las vías Wnt pueden aumentar la proliferación celular. Las investigaciones actuales se encuentran centradas en la acción de las vías Wnt en la regulación de la elección de las células madres entre proliferar o autorenovarse. Esta acción de las vías Wnt en el posible control y mantenimiento de las células madre, puede proveer un posible tratamiento para los tipos de cáncer que exhiben un comportamiento Wnt aberrante.[32][33][34]
Superfamilia TGF-β
Los “Factores de Crecimiento Transformantes” (TGF-Transforming Growth Factor) son una familia de proteínas que comprende a 33 miembros, cada uno de los cuales es un polipéptido dimérico secretado que regulan el desarrollo.[35] Muchos procesos de desarrollo se encuentran bajo su control, entre ellos la gastrulación, simetría axial del organismo, morfogénesis de los órganos, y homeostasis de los tejidos adultos.[36] Todos los ligandos TGF-β se unen ya sea a receptores de tipo I o tipo II, para crear complejos heterotetraméricos.[37]
Vía TGF-β
La vía de señalización TGB beta regula muchos procesos celulares que intervienen en el desarrollo del embrión y en el funcionamiento del organismo adulto, incluyendo crecimiento celular, diferenciación, apoptosis, y homeostasis. Existen cinco tipos de receptores de tipo II y siete receptores de tipo I tanto en humanos como en otros mamíferos. Estos receptores se conocen como "quinasas de especificidad dual" debido a que su dominio quinasa citoplasmático posee una actividad tirosina quinasa débil, pero una potente actividad quinasa de serina/treonina.[38] Cuando un ligando de la superfamilia TGB-β se une a un receptor de tipo II, este recluta a los receptores de tipo I y los activa fosforilando los residuos serina y treonina en su caja "GS".[39] Esto produce un complejo d activación que puede luego fosforilar a las proteínas SMAD por medio de fosrorilación.
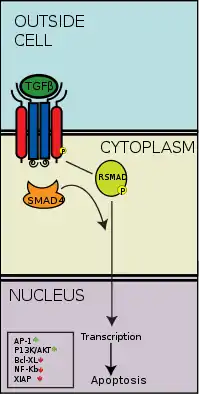
Vía SMAD
Existen tres clases de SMADs:
- SMADs reguladas por receptor (R-SMAD)
- SMADs de mediador común (Co-SMAD)
- SMADs inhibitorias (I-SMAD)
Ejemplos de SMADs de cada clase:[40][41][42]
| Class | SMADs |
|---|---|
| R-SMAD | SMAD1, SMAD2, SMAD3, SMAD5 y SMAD8/9 |
| Co-SMAD | SMAD4 |
| I-SMAD | SMAD6 y SMAD7 |
La superfamilia TGF-β activa a los miembros de la familia SMAD, las cuales funcionan como factores de transcripción. Específicamente, el receptor tipo I, activado por el receptor tipo II, fosforila a las R-SMADs que posteriormente se une a la co-SMAD, SMAD4. Las R-SMAD/Co-SMAD forman un complejo con la importina e ingresa al núcleo, donde actúan como factores de transcripción ya sea regulando al alza o a la baja la expresión de sus genes objetivo.
Algunos ligandos TGF-β específicos desembocan en la activación ya sea de las R-SMADs SMAD2/3 o de las SMAD1/5. Por ejemplo, cuando la activina, NODAL, o TGF-β se unen a los receptores, el complejo receptor fosforilado es capaz de activar a la SMAD2 y SMAD3 fosforilándolas. Sin embargo, cuando un ligando BMP se une a su receptor, el complejo receptor fosforilado activa a la SMAD1 y la SMAD5. Luego los complejos SMAD 2/3 o SMAD 1/5 forman un dímero complejo con SMAD4 y se convierten en factores de transcripción. Aunque hay muchos R-SMADs involucrados en la vía, solo existe un co-SMAD, el SMAD4.[43]
Ejemplos
Los factores de crecimiento y factores de coagulación son agentes de señalización paracrinos. La acción local de la señalización de los factores de crecimiento desempeña un papel especialmente importante en el desarrollo de los tejidos. Además el ácido retinoico, la forma activa de la vitamina A; actúa en forma de factor paracrino regulando la expresión de genes durante el desarrollo embrional en los animales superiores.[44] Las citokinas son un grupo de proteínas que inician la respuesta inflamatoria, usualmente como resultado de una infección. Generalmente inducen la secreción de otras citokinas, moléculas gaseosas de señalización como el óxido nítrico (NO), prostaglandinas y Leucotrienos. Entre las principales citokinas proinflamatorias se encuentran el factor de necrosis tumoral (TNF-a), las interleucinas IL-1, IL-6 e IL-8) y los Interferones.[45]
En los insectos, la allostatina controla el crecimiento por medio de una acción parácrina en la corpora allata.[cita requerida]
En los organismos maduros, la señalización paracrina se encuentra involucrada en las respuestas a alérgenos, reparación de tejidos, formación de tejido cicatrizal, y coagulación de la sangre.[cita requerida]
Véase también
Referencias
- Gospodarowicz, D.; Ferrara, N.; Schweigerer, L.; Neufeld, G. (1987). «Structural Characterization and Biological Functions of Fibroblast Growth Factor». Endocrine Reviews 8 (2): 95-114. PMID 2440668. doi:10.1210/edrv-8-2-95.
- Rifkin, Daniel B.; Moscatelli, David (1989). «Recent developments in the cell biology of basic fibroblast growth factor». The Journal of Cell Biology 109 (1): 1-6. JSTOR 1613457. PMC 2115467. PMID 2545723. doi:10.1083/jcb.109.1.1.
- Lappi, Douglas A. (1995). «Tumor targeting through fibroblast growth factor receptors». Seminars in Cancer Biology 6 (5): 279-88. PMID 8562905. doi:10.1006/scbi.1995.0036.
- Xu, J.; Xu, J; Colvin, JS; McEwen, DG; MacArthur, CA; Coulier, F; Gao, G; Goldfarb, M (1996). «Receptor Specificity of the Fibroblast Growth Factor Family». Journal of Biological Chemistry 271 (25): 15292-7. PMID 8663044. doi:10.1074/jbc.271.25.15292.
- Logan, M. (2003). «Finger or toe: The molecular basis of limb identity». Development 130 (26): 6401-10. PMID 14660539. doi:10.1242/dev.00956.
- Fantl, Wendy J; Johnson, Daniel E; Williams, Lewis T (1993). «Signaling by Receptor Tyrosine Kinases». Annual Review of Biochemistry 62: 453-81. PMID 7688944. doi:10.1146/annurev.bi.62.070193.002321.
- Yarden, Yosef; Ullrich, Axel (1988). «Growth Factor Receptor Tyrosine Kinases». Annual Review of Biochemistry 57: 443-78. PMID 3052279. doi:10.1146/annurev.bi.57.070188.002303.
- Katz, Michael E; McCormick, Frank (1997). «Signal transduction from multiple Ras effectors». Current Opinion in Genetics & Development 7 (1): 75-9. PMID 9024640. doi:10.1016/S0959-437X(97)80112-8.
- Zsebo, Krisztina M.; Williams, David A.; Geissler, Edwin N.; Broudy, Virginia C.; Martin, Francis H.; Atkins, Harry L.; Hsu, Rou-Yin; Birkett, Neal C.; Okino, Kenneth H.; Murdock, Douglas C.; Jacobsen, Frederick W.; Langley, Keith E.; Smith, Kent A.; Takeish, Takashi; Cattanach, Bruce M.; Galli, Stephen J.; Suggs, Sidney V. (1990). «Stem cell factor is encoded at the SI locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor». Cell 63 (1): 213-24. PMID 1698556. doi:10.1016/0092-8674(90)90302-U.
- Rönnstrand, L. (2004). «Signal transduction via the stem cell factor receptor/c-Kit». Cellular and Molecular Life Sciences 61 (19–20): 2535-48. PMID 15526160. doi:10.1007/s00018-004-4189-6.
- Melillo, Rosa Marina; Castellone, Maria Domenica; Guarino, Valentina; De Falco, Valentina; Cirafici, Anna Maria; Salvatore, Giuliana; Caiazzo, Fiorina; Basolo, Fulvio; Giannini, Riccardo; Kruhoffer, Mogens; Orntoft, Torben; Fusco, Alfredo; Santoro, Massimo (2005). «The RET/PTC-RAS-BRAF linear signaling cascade mediates the motile and mitogenic phenotype of thyroid cancer cells». Journal of Clinical Investigation 115 (4): 1068-81. PMC 1062891. PMID 15761501. doi:10.1172/JCI22758.
- Kolch, Walter (2000). «Meaningful relationships: The regulation of the Ras/Raf/MEK/ERK pathway by protein interactions». The Biochemical Journal 351 (2): 289-305. PMC 1221363. PMID 11023813. doi:10.1042/0264-6021:3510289.
- Aaronson, David S.; Horvath, Curt M. (2002). «A Road Map for Those Who Don't Know JAK-STAT». Science 296 (5573): 1653-5. Bibcode:2002Sci...296.1653A. PMID 12040185. doi:10.1126/science.1071545.
- Rawlings, Jason S.; Rosler, Kristin M.; Harrison, Douglas A. (2004). «The JAK/STAT signaling pathway». Journal of Cell Science 117 (8): 1281-3. PMID 15020666. doi:10.1242/jcs.00963.
- O'Shea, John J; Gadina, Massimo; Schreiber, Robert D (2002). «Cytokine signaling in 2002: new surprises in the Jak/Stat pathway». Cell 109 (2): S121-31. PMID 11983158. doi:10.1016/S0092-8674(02)00701-8.
- Shiang, Rita; Thompson, Leslie M.; Zhu, Ya-Zhen; Church, Deanna M.; Fielder, Thomas J.; Bocian, Maureen; Winokur, Sara T.; Wasmuth, John J. (1994). «Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia». Cell 78 (2): 335-42. PMID 7913883. doi:10.1016/0092-8674(94)90302-6.
- Kalluri, Raghu; Weinberg, Robert A. (2009). «The basics of epithelial-mesenchymal transition». Journal of Clinical Investigation 119 (6): 1420-8. PMC 2689101. PMID 19487818. doi:10.1172/JCI39104.
- Silver, Debra L.; Montell, Denise J. (2001). «Paracrine Signaling through the JAK/STAT Pathway Activates Invasive Behavior of Ovarian Epithelial Cells in Drosophila». Cell 107 (7): 831-41. PMID 11779460. doi:10.1016/S0092-8674(01)00607-9.
- Ingham, P. W.; McMahon, AP (2001). «Hedgehog signaling in animal development: Paradigms and principles». Genes & Development 15 (23): 3059-87. PMID 11731473. doi:10.1101/gad.938601.
- Bitgood, Mark J.; McMahon, Andrew P. (1995). «Hedgehog and Bmp Genes Are Coexpressed at Many Diverse Sites of Cell–Cell Interaction in the Mouse Embryo». Developmental Biology 172 (1): 126-38. PMID 7589793. doi:10.1006/dbio.1995.0010.
- Jacob, L.; Lum, L. (2007). «Hedgehog Signaling Pathway». Science's STKE 2007 (407): cm6. PMID 17925577. doi:10.1126/stke.4072007cm6.
- Johnson, Ronald L; Scott, Matthew P (1998). «New players and puzzles in the Hedgehog signaling pathway». Current Opinion in Genetics & Development 8 (4): 450-6. PMID 9729722. doi:10.1016/S0959-437X(98)80117-2.
- Nybakken, K; Perrimon, N (2002). «Hedgehog signal transduction: Recent findings». Current Opinion in Genetics & Development 12 (5): 503-11. PMID 12200154. doi:10.1016/S0959-437X(02)00333-7.
- Collins, R. T.; Cohen, SM (2005). «A Genetic Screen in Drosophila for Identifying Novel Components of the Hedgehog Signaling Pathway». Genetics 170 (1): 173-84. PMC 1449730. PMID 15744048. doi:10.1534/genetics.104.039420.
- Evangelista, M.; Tian, H.; De Sauvage, F. J. (2006). «The Hedgehog Signaling Pathway in Cancer». Clinical Cancer Research 12 (20): 5924-8. PMID 17062662. doi:10.1158/1078-0432.CCR-06-1736.
- Taipale, Jussi; Beachy, Philip A. (2001). «The Hedgehog and Wnt signaling pathways in cancer». Nature 411 (6835): 349-54. PMID 11357142. doi:10.1038/35077219.
- Cadigan, K. M.; Nusse, R. (1997). «Wnt signaling: A common theme in animal development». Genes & Development 11 (24): 3286-305. PMID 9407023. doi:10.1101/gad.11.24.3286.
- Dale, Trevor C. (1998). «Signal transduction by the Wnt family of ligands». The Biochemical Journal 329 (Pt 2): 209-23. PMC 1219034. PMID 9425102. doi:10.1042/bj3290209.
- Chen, Xi; Yang, Jun; Evans, Paul M; Liu, Chunming (2008). «Wnt signaling: The good and the bad». Acta Biochimica et Biophysica Sinica 40 (7): 577-94. PMC 2532600. PMID 18604449. doi:10.1111/j.1745-7270.2008.00440.x.
- Komiya, Yuko; Habas, Raymond (2008). «Wnt signal transduction pathways». Organogenesis 4 (2): 68-75. PMC 2634250. PMID 19279717. doi:10.4161/org.4.2.5851.
- Logan, Catriona Y.; Nusse, Roel (2004). «The Wnt Signaling Pathway in Development and Disease». Annual Review of Cell and Developmental Biology 20: 781-810. PMID 15473860. doi:10.1146/annurev.cellbio.20.010403.113126.
- Lustig, B; Behrens, J (2003). «The Wnt signaling pathway and its role in tumor development». Journal of cancer research and clinical oncology 129 (4): 199-221. PMID 12707770. doi:10.1007/s00432-003-0431-0.
- Neth, Peter; Ries, Christian; Karow, Marisa; Egea, Virginia; Ilmer, Matthias; Jochum, Marianne (2007). «The Wnt Signal Transduction Pathway in Stem Cells and Cancer Cells: Influence on Cellular Invasion». Stem Cell Reviews 3 (1): 18-29. PMID 17873378. doi:10.1007/s12015-007-0001-y.
- Bandyopadhyay, Amitabha; Tsuji, Kunikazu; Cox, Karen; Harfe, Brian D.; Rosen, Vicki; Tabin, Clifford J. (2006). «Genetic Analysis of the Roles of BMP2, BMP4, and BMP7 in Limb Patterning and Skeletogenesis». PLoS Genetics 2 (12): e216. PMC 1713256. PMID 17194222. doi:10.1371/journal.pgen.0020216.
- Attisano, Liliana; Wrana, Jeffrey L. (2002). «Signal Transduction by the TGF-β Superfamily». Science 296 (5573): 1646-7. Bibcode:2002Sci...296.1646A. PMID 12040180. doi:10.1126/science.1071809.
- Wrana, Jeffrey L.; Ozdamar, Barish; Le Roy, Christine; Benchabane, Hassina (2008). «Signaling Receptors of the TGF-β Family». En Derynck, Rik; Miyazono, Kohei, eds. The TGF-β Family. pp. 151-77. ISBN 978-0-87969-752-5.
- ten Dijke, Peter; Heldin, Carl-Henrik (2006). «The Smad family». En ten Dijke, Peter; Heldin, Carl-Henrik, eds. Smad Signal Transduction: Smads in Proliferation, Differentiation and Disease. Proteins and Cell Regulation 5. Dordrecht: Springer. pp. 1-13. ISBN 978-1-4020-4709-1.
- Moustakas, Aristidis (1 de septiembre de 2002). «Smad signaling network». Journal of Cell Science 115 (17): 3355-6. PMID 12154066.
- Wu, Jia-Wei; Hu, Min; Chai, Jijie; Seoane, Joan; Huse, Morgan; Li, Carey; Rigotti, Daniel J.; Kyin, Saw; Muir, Tom W.; Fairman, Robert; Massagué, Joan; Shi, Yigong (2001). «Crystal Structure of a Phosphorylated Smad2». Molecular Cell 8 (6): 1277-89. PMID 11779503. doi:10.1016/S1097-2765(01)00421-X.
- Pavletich, Nikola P.; Hata, Yigong; Lo, Akiko; Massagué, Roger S.; Pavletich, Joan (1997). «A structural basis for mutational inactivation of the tumour suppressor Smad4». Nature 388 (6637): 87-93. PMID 9214508. doi:10.1038/40431.
- Itoh, Fumiko; Asao, Hironobu; Sugamura, Kazuo; Heldin, Carl-Henrik; Ten Dijke, Peter; Itoh, Susumu (2001). «Promoting bone morphogenetic protein signaling through negative regulation of inhibitory Smads». The EMBO Journal 20 (15): 4132-42. PMC 149146. PMID 11483516. doi:10.1093/emboj/20.15.4132.
- Schmierer, Bernhard; Hill, Caroline S. (2007). «TGFβ–SMAD signal transduction: Molecular specificity and functional flexibility». Nature Reviews Molecular Cell Biology 8 (12): 970-82. PMID 18000526. doi:10.1038/nrm2297.
- Duester, Gregg (September 2008). «Retinoic acid synthesis and signaling during early organogenesis». Cell 134 (6): 921-31. PMC 2632951. PMID 18805086. doi:10.1016/j.cell.2008.09.002.
- «Fisiología de la Inflamación». Sociedad Argentina de Terapia Intensiva. 25 de septiembre de 2006. Archivado desde el original el 30 de septiembre de 2007. Consultado el 23 de mayo de 2020.
Enlaces externos
- MeSH: Paracrine+Signaling (en inglés)
- paracrine en el Diccionario Médico de Dorland
 Datos: Q14873666
Datos: Q14873666 Multimedia: Paracrine communication / Q14873666
Multimedia: Paracrine communication / Q14873666