Chimie hôte-invité
En chimie supramoléculaire[1], la chimie hôte-invité décrit des complexes qui sont composés de deux ou plusieurs molécules ou ions qui sont maintenus ensemble par des forces autres que celles des liaisons covalentes. La chimie hôte-invité englobe l'idée de la reconnaissance moléculaire et des interactions par liaison non-covalente. Cette dernière est essentielle pour maintenir la structure 3D de grandes molécules, telles que les protéines, et est impliquée dans de nombreux processus biologiques dans lesquels de grandes molécules se lient spécifiquement mais de manière transitoire les unes aux autres.
Bien que les interactions non-covalentes puissent être grossièrement divisées en celles ayant des contributions plus électrostatiques ou dispersives, il existe d'autres types d'interactions non-covalentes : liaison ionique, liaison hydrogène, forces de van der Waals et interactions hydrophobes[2].
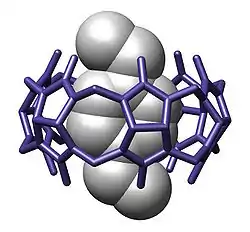
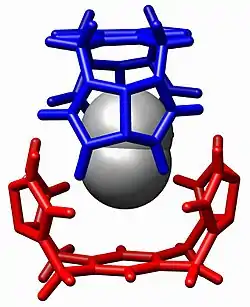
Aperçu
La chimie hôte-invité est une branche de la chimie supramoléculaire dans laquelle une molécule hôte forme un composé chimique avec une molécule ou un ion invité. Les deux composants du complexe sont maintenus ensemble par des forces non-covalentes, le plus souvent par liaison hydrogène. La liaison entre l'hôte et l'invité est généralement très spécifique aux deux parties concernées. La formation de ces complexes est au cœur du sujet de la reconnaissance moléculaire.
Il existe un équilibre entre l'état non lié, dans lequel l'hôte et l'invité sont séparés l'un de l'autre, et l'état lié, dans lequel ils forment un complexe hôte-invité défini structurellement :
- H = « hôte », G = « invité », HG = « complexe hôte-invité »

Le composant « hôte » peut être considéré comme la plus grande molécule, et il englobe la plus petite molécule « invitée ». Dans les systèmes biologiques, les termes analogues d'hôte et d'invité sont communément appelés « enzyme » et « substrat »[5].
Afin de concevoir des systèmes synthétiques qui exécutent des fonctions et des tâches spécifiques, il est très important de comprendre la thermodynamique de la liaison entre l'hôte et l'invité. Les chimistes se concentrent sur l'échange d'énergie de différentes interactions de liaison et tentent de développer des expériences scientifiques pour quantifier les origines fondamentales de ces interactions non-covalentes en utilisant diverses techniques telles que la spectroscopie RMN, la spectroscopie ultraviolet-visible et la calorimétrie de titrage isotherme[6]. L'analyse quantitative des valeurs constantes de liaison fournit des informations thermodynamiques utiles[5].
Principes thermodynamiques des interactions hôte-invité
Les avantages thermodynamiques de la chimie hôte-invité viennent de l'idée qu'il existe une énergie libre de Gibbs globale plus faible en raison de l'interaction entre les molécules hôtes et invitées. Les chimistes tentent de mesurer l'énergie et les propriétés thermodynamiques de ces interactions non-covalentes; et ce faisant, nous espérons avoir un meilleur aperçu du résultat combinatoire de ces nombreuses petites forces non-covalentes qui sont utilisées pour générer un effet global sur la structure supramoléculaire.
Une constante d'association peut être définie par l'expression


où {HG} est l'activité thermodynamique du complexe à l'équilibre. {H} représente l'activité de l'hôte et {G} représente l'activité de l'invité. Les quantités , et sont les concentrations et est un quotient des coefficients d'activité.




En pratique, la constante d'équilibre est généralement définie en termes de concentrations :

Lorsque cette définition est utilisée, il est sous-entendu que le quotient des coefficients d'activité a une valeur numérique de 1. Il apparaît alors que la constante d'équilibre a la dimension 1/concentration, mais cela ne peut pas être vrai puisque le changement d'énergie libre standard de Gibbs, est proportionnel au logarithme de K.



Ce paradoxe est résolu lorsque la dimension de est définie comme l'inverse de la dimension du quotient des concentrations. Le facteur est considéré comme ayant une valeur constante dans toutes les conditions expérimentales pertinentes. Néanmoins, il est courant d'attacher une dimension, telle que millimole par litre ou micromole par litre, à une valeur de K qui a été déterminée expérimentalement.
Une grande valeur de indique que les molécules hôtes et invitées interagissent fortement pour former le complexe hôte-invité.
Détermination des valeurs de constantes de liaison
Complexation simple hôte-invité
Lorsque les molécules hôtes et invitées se combinent pour former un seul complexe, l'équilibre est représenté par

et la constante d'équilibre, K, est définie comme

où [X] désigne la concentration d'une espèce chimique X (tous les coefficients d'activité sont supposés avoir une valeur numérique de 1). Les équations, à tout point de données,


où et représentent les concentrations totales de l'hôte et de l'invité, peuvent être réduites à une seule équation quadratique et, par exemple, [G] et peuvent donc être résolues analytiquement pour toute valeur donnée de K. Les concentrations [H] et [HG] peuvent alors être dérivées.




La prochaine étape consiste à calculer la valeur, , d'une quantité correspondant à la quantité observée . Ensuite, une somme de carrés, U, sur tous les points de données, np, peut être définie comme



et cela peut être minimisé par rapport à la valeur de constante de stabilité, K, et à un paramètre tel que le déplacement chimique du complexe HG (données RMN) ou son absorbance molaire (données UV/visible). La minimisation peut être effectuée dans une application de feuille de calcul telle qu'Excel à l'aide de l'utilitaire Solver intégré.
Cette procédure ne doit être utilisée que lorsqu'il est certain que l'adduit 1:1 est la seule espèce complexe formée. Un simple contrôle de la validité de cette assertion est que les résidus devraient montrer une distribution aléatoire ; sinon, la formation d'une seconde espèce doit être envisagée.

Calorimétrie
La chaleur dégagée lorsqu'une aliquote de solution hôte est ajoutée à une solution contenant l'invité est la somme des contributions de chaque réaction telle que

où est une valeur de changement de chaleur mesurée (corrigée pour toutes les contributions de chaleur étrangères) au point de données j. est la quantité de chaleur absorbée ou émise lorsqu'une mole du ième produit de réaction est formée et est le changement réel du nombre de moles de ce produit à ce point de données. est calculé en résolvant les équations du bilan massique avec des valeurs données des constantes d'équilibre. Si les valeurs de la constante d'équilibre sont connues, le changement d'enthalpie standard peut être calculé par un processus linéaire des moindres carrés, sinon une méthode non linéaire d'ajustement des données doit être utilisée.




La calorimétrie par titrage isotherme est couramment utilisée pour déterminer les valeurs à la fois d'une constante d'équilibre et de l'enthalpie de réaction standard correspondante. Les fabricants d'instruments fournissent certains logiciels avec lesquels ces quantités peuvent être obtenues à partir de valeurs de données expérimentales.
Réaction de complexation générale
Pour chaque équilibre impliquant un hôte, H, et un invité G :

la constante d'équilibre, , est définie comme


Les valeurs des concentrations libres et sont obtenus en résolvant les équations avec des valeurs connues ou estimées pour les constantes de stabilité.


Ensuite, les concentrations de chaque espèce complexe peuvent également être calculées comme . La relation entre la concentration d'une espèce et la quantité mesurée est spécifique à la technique de mesure, comme indiqué dans chaque section ci-dessus. En utilisant cette relation, l'ensemble de paramètres, les valeurs de constantes de stabilité et les valeurs de propriétés telles que l'absorbance molaire ou les déplacements chimiques spécifiés, peuvent être affinés par un processus de raffinement non linéaire des moindres carrés. Pour une présentation plus détaillée de la théorie, voir Détermination des constantes d'équilibre. Certains programmes informatiques dédiés sont répertoriés dans des logiciels.

Techniques expérimentales
Résonance magnétique nucléaire
La résonance magnétique nucléaire (RMN) est l'une des techniques spectroscopiques les plus puissantes en chimie analytique. C'est un outil important pour l'étude des complexes hôte-invité, pour élucider les structures des différents complexes existant sous forme d'agrégats, de paires d'ions ou de systèmes encapsulés. Comme son nom l'indique, la RMN identifie les différents noyaux des molécules (le plus souvent, le proton), en mesurant leur déplacement chimique. L'association entre deux molécules entraîne une modification considérable de leurs environnements électroniques. Cela conduit à un déplacement des signaux dans le spectre RMN, et ce principe de base est utilisé pour étudier les phénomènes de chimie hôte-invité. Les forces motrices de la liaison hôte-invité sont les diverses interactions secondaires entre les molécules, telles que la liaison hydrogène et l'interaction pi. Ainsi, la RMN sert également de technique importante pour établir la présence de ces interactions dans un complexe hôte-invité[7].

Des études de RMN ont donné des informations utiles sur la liaison de différents invités aux hôtes. Fox et al.[8] ont calculé les interactions de liaison hydrogène entre les molécules de pyridine et de dendrimère poly(amidoamine) (PAMAM), sur la base du déplacement chimique de l'amine et des groupes amide. Dans une étude similaire, Xu et al.[9] titré le dendrimère G4 PAMAM à base de carboxylate (l'hôte) avec divers médicaments à base d'amine (les invités) et surveillé les déplacements chimiques du dendrimère. En conjonction avec les techniques de RMN 2D-NOESY, ils ont pu localiser précisément la position des médicaments sur les dendrimères et l'effet de la fonctionnalité sur l'affinité de liaison des médicaments. Ils ont trouvé des preuves concluantes pour montrer que les molécules de médicaments cationiques se fixent à la surface des dendrimères anioniques par des interactions électrostatiques, alors qu'un médicament anionique se localise à la fois dans le noyau et la surface des dendrimères, et que la force de ces interactions dépend des valeurs de pKa des molécules.
Dans une autre étude, Sun et al.[9] ont étudié la chimie hôte-invité des molécules de ruthénium trisbipyridyl-viologène avec le cucurbiturile. Tout en étudiant le changement des déplacements chimiques des protons de pyridine sur le viologène, ils ont constaté que les modes de liaison pour les complexes 1:1 sont complètement différents pour les différentes molécules de cucurbiturile.
Un facteur important à garder à l'esprit lors de l'analyse de la liaison entre l'hôte et l'invité est le temps nécessaire à l'acquisition des données par rapport au temps de l'événement de liaison. Dans de nombreux cas, les événements de liaison sont beaucoup plus rapides que l'échelle de temps d'acquisition des données, auquel cas le résultat est un signal moyenné pour les molécules individuelles et le complexe. L'échelle de temps RMN est de l'ordre de la milliseconde, ce qui dans certains cas lorsque la réaction de liaison est rapide, limite la précision de la technique[5].
Spectroscopie ultraviolet-visible

La spectroscopie ultraviolet-visible est l'une des méthodes les plus anciennes et les plus rapides pour étudier l'activité de liaison de diverses molécules. L'absorption de la lumière UV a lieu à une échelle de temps de 1 × 10−12 s (une picoseconde), d'où les signaux individuels de l'espèce peuvent être observés. Dans le même temps, l'intensité de l'absorption est directement corrélée à la concentration de l'espèce, ce qui permet un calcul facile de la constante d'association[5]. Le plus souvent, l'hôte ou l'invité est transparent aux rayons UV, tandis que l'autre molécule est sensible aux UV. L'évolution de la concentration des molécules sensibles aux UV est ainsi suivie et ajustée en ligne droite par la méthode de Benesi-Hildebrand (en), à partir de laquelle la constante d'association peut être directement calculée.
Des informations supplémentaires sur la stœchiométrie des complexes sont également obtenues, car la méthode de Benesi-Hildebrand suppose une stœchiométrie 1:1 entre l'hôte et l'invité. Les données traceront une ligne droite seulement si la formation complexe suit également une stœchiométrie 1:1 similaire. Un exemple récent d'un calcul similaire a été effectué par Sun et al.[9], dans lequel ils ont titré des molécules de ruthénium trisbipyridyl-viologène avec des urilles de cucurbitacées. Ils ont tracé l'absorbance relative des molécules de cucurbitacées en fonction de leur concentration totale à une longueur d'onde spécifique. Les données ont bien adapté un modèle de liaison 1:1 avec une constante de liaison de 1,2 × 105 M−1.
En tant qu'extension, on peut adapter les données à différentes stœchiométrie pour comprendre la cinétique de liaison entre l'hôte et l'invité[10] se sont servis de ce corollaire pour modifier légèrement le tracé Benesi-Hildebrand conventionnel pour obtenir l'ordre de la réaction de complexation entre le complexe hétérotrinucléaire salen (en) Zn(II) chiral ponté par l'éther couronne contenant du baryum (hôte) avec divers invités imidazoles et esters méthyliques d'acides aminés, ainsi que les autres paramètres. Ils ont titré une concentration fixe du complexe de zinc avec des quantités variables d'imidazoles et d'esters méthyliques tout en surveillant les changements de l'absorbance de la bande de transition pi-pi* à 368 nm. Les données correspondent à un modèle dans lequel le rapport invité-hôte est de 2 dans le complexe. Ils ont ensuite mené ces expériences à différentes températures qui leur ont permis de calculer les différents paramètres thermodynamiques à l'aide de l'équation de van 't Hoff.

Calorimétrie par titrage isotherme
Les techniques spectroscopiques donnent les valeurs de la constante de liaison et l'énergie libre de Gibbs, . Pour obtenir l'ensemble complet des paramètres thermodynamiques tels que et , une analyse de van 't Hoff utilisant l'équation de van 't Hoff serait nécessaire. Cependant, les progrès récents des techniques calorimétriques permettent de mesurer et en une seule expérience, permettant ainsi de déterminer tous les paramètres thermodynamiques à l'aide de l'équation :




à condition que l'expérience soit réalisée dans des conditions isothermes. La procédure est similaire à une procédure de titrage conventionnelle dans laquelle l'hôte est ajouté séquentiellement à l'invité et la chaleur absorbée ou dégagée est mesurée, comparée à une solution à blanc. La chaleur totale dégagée, Q, correspond à la constante d'association, , et par l'équation :


Ce qui peut être simplifié comme

où
- : concentration molaire initiale de l'hôte,
- : concentration molaire de l'invité,
- : volume du récipient.


L'équation ci-dessus peut être résolue par une analyse de régression non linéaire pour obtenir les valeurs de et et par la suite de et pour cette réaction particulière[5]. Les avantages de la calorimétrie par titrage isotherme par rapport aux autres techniques couramment utilisées, outre le fait de donner l'ensemble des paramètres thermodynamiques, sont qu'elle est plus générale et adaptée à une large gamme de molécules. Il n'est pas nécessaire d'avoir des composés avec des chromophores ou des groupes fonctionnels UV-visibles car le signal thermique est une propriété universelle des réactions de liaison. Dans le même temps, le rapport signal sur bruit est assez favorable, ce qui permet une détermination plus précise des constantes de liaison, même dans des conditions très diluées[11]. Un exemple récent de l'utilisation de cette technique était d'étudier l'affinité de liaison de la membrane protéique entourant Escherichia coli aux cations lipophiles utilisés dans les médicaments dans divers environnements mimétiques membranaires. La motivation de l'étude ci-dessus était que ces membranes rendent les bactéries résistantes à la plupart des composés à base de cation ammonium quaternaire, qui ont des effets antibactériens. Ainsi, une compréhension des phénomènes de liaison permettrait de concevoir des antibiotiques efficaces contre E. coli. Les chercheurs ont maintenu un large excès de ligand par rapport à la protéine pour permettre à la réaction de liaison de se terminer. En utilisant les équations ci-dessus, les chercheurs ont procédé au calcul , , et pour chaque médicament dans des environnements différents. Les données ont indiqué que la stœchiométrie de liaison du médicament avec la membrane était de 1:1 avec une valeur micromolaire de . Les valeurs négatives de , et ont indiqué que le processus était entraîné par l'enthalpie avec une valeur de 8 à 12 kcal/mol pour chaque médicament[12].
Applications
Spectroscopie Raman
La spectroscopie Raman est une technique spectroscopique utilisée dans l'étude des molécules qui présentent un effet de diffusion Raman lorsqu'elles sont irradiées en lumière monochromatique incidente. La condition de base pour obtenir un signal Raman est que la lumière incidente provoque une transition électronique dans l'espèce chimique de son état fondamental à un état d'énergie virtuelle, qui émettra un photon au retour à l'état fondamental. La différence d'énergie entre le photon absorbé et émis est unique pour chaque espèce chimique en fonction de son environnement électronique. Par conséquent, la technique sert d'outil important pour l'étude de divers événements de liaison, car la liaison entre les molécules entraîne presque toujours un changement dans leur environnement électronique. Cependant, ce qui fait de la spectroscopie Raman une technique unique, c'est que seules les transitions accompagnées d'un changement de polarisation de la molécule sont actives Raman. Les informations structurelles dérivées des spectres Raman donnent des informations très spécifiques sur la configuration électronique du complexe par rapport à l'hôte individuel et aux molécules invitées.
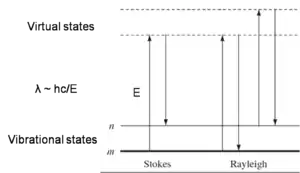
La spectroscopie Raman en phase solution aboutit souvent à une faible diffusion. Par conséquent, des progrès récents ont été réalisés pour améliorer les signaux Raman, tels que la spectroscopie Raman à surface améliorée et la spectroscopie Raman par résonance. Ces techniques servent à quantifier les liaisons analyte-récepteur, donnant une image plus détaillée des phénomènes de complexation hôte-invité où ils se produisent réellement, dans les solutions. Dans une percée récente, Flood et al. ont déterminé la force de liaison du tétrathiafulvalène (TTF) et du cyclobis (paraquat-p-phénylène) en utilisant la spectroscopie Raman[13] ainsi que le SERS[14]. Les travaux antérieurs dans ce domaine visaient à fournir des informations sur la liaison et la structure du complexe résultant, plutôt que des mesures quantitatives des forces d'association. Les chercheurs ont dû utiliser la spectroscopie Raman par résonance afin de pouvoir obtenir des signaux détectables à partir de solutions avec des concentrations aussi faibles que 1 mM. En particulier, ils ont corrélé l'intensité des bandes Raman avec la géométrie du complexe à l'état photo-excité. Semblable au titrage basé sur la spectroscopie ultraviolet-visible, ils ont calculé la constante de liaison par « titrage Raman » et ajusté les courbes de liaison à des modèles 1:1, donnant un de −5,7 ± 0,6 kcal/mol. L'étude fournit maintenant une base pour des études similaires impliquant des complexes de transfert de charge dans des solutions.
Coopérativité
La coopérativité est définie comme étant lorsqu'un ligand se lie à un récepteur sur plusieurs sites de liaison, le ligand provoque une diminution ou une augmentation de l'affinité pour les ligands entrants. S'il y a une augmentation de la liaison des ligands suivants, cela est considéré comme une coopérativité positive. Si une diminution de la liaison est observée, il s'agit d'une coopérativité négative. Des exemples de coopérativité positive et négative sont l'hémoglobine et le récepteur d'aspartate, respectivement[15].
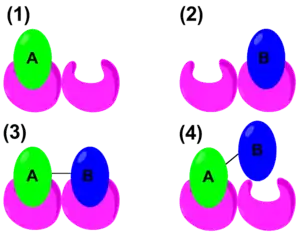
Ces dernières années, les propriétés thermodynamiques de la coopérativité ont été étudiées afin de définir des paramètres mathématiques qui distinguent la coopérativité positive ou négative. L'équation traditionnelle de l'énergie libre de Gibbs est : . Cependant, pour quantifier la coopérativité dans un système hôte-invité, l'énergie de liaison doit être prise en compte. Le schéma de droite montre la liaison de A, la liaison de B, la liaison coopérative positive de A – B, et enfin, la liaison coopérative négative de A – B. Par conséquent, une forme alternative de l'équation d'énergie libre de Gibbs serait



où :
- : énergie libre de liaison A ;
- : énergie libre de liaison B ;
- : énergie libre de liaison pour A et B attachés ;
- : somme des énergies libres de liaison.




On considère que si est plus que la somme de et , alors il est positivement coopératif. Si est moins, alors il est négativement coopératif[16]. La chimie hôte-invité ne se limite pas aux interactions récepteur-ligand. Il est également démontré dans les systèmes d'appariement d'ions. Ces dernières années, de telles interactions sont étudiées dans un milieu aqueux utilisant des hôtes organométalliques et des molécules organiques hôtes. Par exemple, un récepteur poly-cationique contenant du cuivre (l'hôte) est coordonné avec des molécules telles que les tétracarboxylates, le tricarballate, l'aspartate et l'acétate (les invités). Cette étude illustre que l'entropie plutôt que l'enthalpie détermine l'énergie de liaison du système conduisant à une coopérativité négative. Le grand changement d'entropie provient du déplacement des molécules de solvant entourant le ligand et le récepteur. Lorsque plusieurs acétates se lient au récepteur, il libère plus de molécules d'eau dans l'environnement qu'un tétracarboxylate. Cela a conduit à une diminution de l'énergie libre, ce qui implique que le système coopère négativement[17]. Dans une étude similaire, utilisant du guanidinium et du Cu(II) et des invités polycarboxylates, il est démontré que le positif en coopération est largement déterminé par l'enthalpie[18]. En plus des études thermodynamiques, la chimie hôte-invité a également des applications biologiques.
Supraconductivité
Aux basses températures et aux pressions élevées, le bismuth présente une structure hôte-invité. Cela conduit étonnamment à une forte supraconductivité de couplage[19].
Application biologique

Les dendrimères dans les systèmes d'administration de médicaments sont un exemple d'interactions hôte-invité. L'interaction entre l'hôte et l'invité, le dendrimère et le médicament, respectivement, peut être hydrophobe ou covalente. L'interaction hydrophobe entre l'hôte et l'invité est considérée comme « encapsulée », tandis que les interactions covalentes sont considérées comme conjuguées. Il a été démontré que l'utilisation de dendrimères en médecine améliore l'administration du médicament en augmentant la solubilité et la biodisponibilité du médicament. En conjonction, les dendrimères peuvent augmenter l'absorption cellulaire et la capacité de ciblage, et diminuer la résistance aux médicaments[20].
La solubilité de divers anti-inflammatoires non stéroïdiens (AINS) augmente lorsqu'ils sont encapsulés dans des dendrimères PAMAM[21]. Cette étude montre que l'amélioration de la solubilité des AINS est due aux interactions électrostatiques entre les groupes amine de surface dans PAMAM et les groupes carboxyle trouvés dans les AINS. Les interactions hydrophobes entre les groupes aromatiques dans les médicaments et les cavités intérieures du dendrimère contribuent à l'augmentation de la solubilité[22]. Lorsqu'un médicament est encapsulé dans un dendrimère, ses propriétés physiques et physiologiques restent inchangées, y compris la non-spécificité et la toxicité. Cependant, lorsque le dendrimère et le médicament sont liés ensemble de manière covalente, il peut être utilisé pour un ciblage tissulaire spécifique et des vitesses de libération contrôlées[23]. La conjugaison covalente de plusieurs médicaments sur les surfaces des dendrimères peut poser un problème d'insolubilité[24].
Ce principe est également à l'étude pour l'application du traitement du cancer. Plusieurs groupes ont encapsulé des médicaments anticancéreux tels que la camptothécine, le méthotrexate et la doxorubicine. Les résultats de ces recherches ont montré que les dendrimères ont une solubilité aqueuse accrue, une vitesse de libération ralentie et peuvent éventuellement contrôler la cytotoxicité des médicaments[20]. Le cisplatine a été conjugué à des dendrimères PAMAM qui ont abouti aux mêmes résultats pharmacologiques que ceux énumérés ci-dessus, mais la conjugaison a également contribué à l'accumulation de cisplatine dans les tumeurs solides lors d'une administration intraveineuse[25].
Détection
Traditionnellement, la détection chimique a été abordée avec un système qui contient un indicateur lié de manière covalente à un récepteur via un lieur. Une fois que l'analyte se lie, l'indicateur change de couleur ou devient fluorescent. Cette technique est appelée « approche indicateur-espaceur-récepteur » (ISR)[26]. Contrairement à l'ISR, le test de déplacement d'indicateur (IDA) utilise une interaction non-covalente entre un récepteur (l'hôte), un indicateur et un analyte (l'invité). Semblable à ISR, l'IDA utilise également des indicateurs colorés (C-IDA) et de fluorescence (F-IDA). Dans un test IDA, un récepteur est incubé avec l'indicateur. Lorsque l'analyte est ajouté au mélange, l'indicateur est rejeté dans l'environnement. Une fois que l'indicateur est relâché, il change de couleur (C-IDA) ou devient fluorescent (F-IDA)[27].

L'IDA offre plusieurs avantages par rapport à l'approche traditionnelle de détection chimique ISR. Premièrement, il ne nécessite pas que l'indicateur soit lié de manière covalente au récepteur. Deuxièmement, puisqu'il n'y a pas de liaison covalente, divers indicateurs peuvent être utilisés avec le même récepteur. Enfin, les milieux dans lesquels le test peut être utilisé sont plus divers[28].
Les techniques de détection chimique telles que le C-IDA ont des implications biologiques. Par exemple, la protamine est un coagulant qui est couramment administré après une chirurgie cardio-pulmonaire qui neutralise l'activité anticoagulante de l'hérapine. Afin de quantifier la protamine dans les échantillons de plasma, un test de déplacement colorimétrique est utilisé. Le colorant Azure A est bleu lorsqu'il n'est pas lié, mais lorsqu'il est lié à l'herapine, il présente une couleur violette. La liaison entre l'Azure A et l'héparine est faible et réversible. Cela permet à la protamine de déplacer l'Azure A. Une fois que le colorant est libéré, il affiche une couleur violette. Le degré de déplacement du colorant est proportionnel à la quantité de protamine dans le plasma[29].
Le F-IDA a été utilisé par Kwalczykowski et ses collègues pour surveiller les activités de l'hélicase dans E. coli. Dans cette étude, ils ont utilisé le thiazole orange comme indicateur. L'hélicase déroule le dsDNA pour faire du ssADN. L'intensité de fluorescence du thiazole orange a une plus grande affinité pour le dsDNA que le ssDNA et son intensité de fluorescence augmente quand il est lié à dsDNA que quand il n'est pas lié[30],[31].
Commutation conformationnelle
Un solide cristallin est considéré comme une entité statique où les mouvements de ses composants atomiques sont limités à son équilibre vibratoire. Comme le montre la transformation du graphite en diamant, la transformation solide en solide peut se produire sous pression physique ou chimique. Il a été récemment proposé que la transformation d'un arrangement cristallin à un autre se fasse de manière coopérative[32],[33]. La plupart de ces études se sont concentrées sur l'étude d'un cadre organique ou métal-organique[34],[35]. En plus des études de transformation cristalline macromoléculaire, il existe également des études de molécules monocristallines qui peuvent changer leur conformation en présence de solvants organiques. Il a été démontré qu'un complexe organométallique se transforme en diverses orientations selon qu'il est exposé ou non à des vapeurs de solvant[36].
Applications environnementales
Des systèmes hôtes-invités ont été utilisés pour éliminer les matières dangereuses pour l'environnement. Ils peuvent être fabriqués dans différentes tailles et différentes formes pour piéger une variété d'invités chimiques. Une application est la capacité du p-tert-butycalix[4]arène à piéger un ion césium. Le césium 137 est radioactif et il est nécessaire de l'éliminer des déchets nucléaires de manière efficace. La chimie hôte-invité a également été utilisée pour éliminer les amines aromatiques cancérigènes et leurs dérivés N-nitroso de l'eau. Ces déchets sont utilisés dans de nombreux processus industriels et se retrouvent dans une variété de produits tels que : les pesticides, les médicaments et les cosmétiques[37],[38].
Références
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Host–guest chemistry » (voir la liste des auteurs).
- Jonathan W. Steed et Jerry L. Atwood, Supramolecular Chemistry, , 2e éd., 1002 p. (ISBN 978-0-470-51234-0).
- Lodish, H., Berk, A. et Kaiser, C., Molecular Cell Biology, (ISBN 978-0-7167-7601-7)
- Freeman, « Structures of the p-xylylenediammonium chloride and calcium hydrogensulfate adducts of the cavitand 'cucurbituril', C36H36N24O12 », Acta Crystallographica B, vol. 40, no 4, , p. 382–387 (DOI 10.1107/S0108768184002354)
- Valdés, Toledo, Spitz et Rebek, « Structure and Selectivity of a Small Dimeric Encapsulating Assembly », Chem. Eur. J., vol. 2, no 8, , p. 989–991 (DOI 10.1002/chem.19960020814)
- Eric V. Anslyn et Dennis A. Dougherty, Modern Physical Organic Chemistry, MacMillan, (ISBN 978-1-891389-31-3)
- Piñeiro, Banquy, Pérez-Casas et Tovar, « On the Characterization of Host–Guest Complexes: Surface Tension, Calorimetry, and Molecular Dynamics of Cyclodextrins with a Non-ionic Surfactant », Journal of Physical Chemistry B, vol. 111, no 17, , p. 4383–92 (PMID 17428087, DOI 10.1021/jp0688815)
- Hu, J, Cheng, Y, Wu, Q et Zhao, L, « Host–Guest Chemistry of Dendrimer-Drug Complexes. 2. Effects of Molecular Properties of Guests and Surface Functionalities of Dendrimers », Journal of Physical Chemistry B, vol. 113, no 31, , p. 10650–10659 (PMID 19603764, DOI 10.1021/jp9047055)
- Santo, M et Fox, M, « Hydrogen bonding interactions between Starburst dendrimers and several molecules of biological interest », Journal of Physical Organic Chemistry, vol. 12, no 4, , p. 293–307 (DOI 10.1002/(SICI)1099-1395(199904)12:4<293::AID-POC88>3.0.CO;2-Q)
- Sun, S, Zhang, R, Andersson, S et Pan, J, « Host–Guest Chemistry and Light Driven Molecular Lock of Ru(bpy)3-Viologen with Cucurbit[7-8]urils », Journal of Physical Chemistry B, vol. 111, no 47, , p. 13357–13363 (PMID 17960929, DOI 10.1021/jp074582j)
- Zhu, Ruan, Chen et Zhang, « Spectroscopy, NMR and DFT studies on molecular recognition of crown ether bridged chiral heterotrinuclear salen Zn(II) complex », Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, vol. 62, nos 4–5, , p. 886–895 (PMID 15897004, DOI 10.1016/j.saa.2005.03.021, Bibcode 2005AcSpA..62..886G)
- Brandts, Williston, Brandts et Lin, « Rapid measurements of Binding Constants and Heats of binding Using a New Titration Calorimeter », Analytical Biochemistry, vol. 179, no 1, , p. 131–137 (PMID 2757186, DOI 10.1016/0003-2697(89)90213-3)
- Sikora, C et Turner, R, « Investigation of Ligand Binding to the Multidrug Resistance Protein EmrE by Isothermal Titration Calorimetry », Biophysical Journal, vol. 88, no 1, , p. 475–482 (PMID 15501941, PMCID 1305024, DOI 10.1529/biophysj.104.049247, Bibcode 2005BpJ....88..475S)
- Witlicki, Hansen, Christensen et Hansen, « Determination of Binding Strengths of a Host–Guest Complex Using Resonance Raman Scattering », Journal of Physical Chemistry A, vol. 113, no 34, , p. 9450–9457 (PMID 19645430, DOI 10.1021/jp905202x, Bibcode 2009JPCA..113.9450W)
- Witlicki, Andersen, Hansen et Jeppesen, « Turning on Resonant SERRS Using the Chromophore-Plasmon Coupling Created by Host–Guest Complexation at a Plasmonic Nanoarray », Journal of the American Chemical Society, vol. 132, no 17, , p. 6099–6107 (PMID 20387841, DOI 10.1021/ja910155b)
- Koshland, D, « The structural basis of negative cooperativity: receptors and enzymes », Current Opinion in Structural Biology, vol. 6, no 6, , p. 757–761 (PMID 8994875, DOI 10.1016/S0959-440X(96)80004-2)
- Jencks, W. P., « On the attribution and additivity of binding energies », Proceedings of the National Academy of Sciences, vol. 78, no 7, , p. 4046–4050 (PMID 16593049, PMCID 319722, DOI 10.1073/pnas.78.7.4046, Bibcode 1981PNAS...78.4046J)
- Dobrzanska, L, Lloyd, G, Esterhuysen, C et Barbour, L, « Studies into the Thermodynamic Origin of Negative Cooperativity in Ion-Pairing Molecular Recognition », Journal of the American Chemical Society, vol. 125, no 36, , p. 10963–10970 (PMID 12952478, DOI 10.1021/ja030265o)
- Hughes, A. et Anslyn, E, « A cationic host displaying positive cooperativity in water », Proceedings of the National Academy of Sciences, vol. 104, no 16, , p. 6538–6543 (PMID 17420472, PMCID 1871821, DOI 10.1073/pnas.0609144104, Bibcode 2007PNAS..104.6538H)
- (en) Brown, Semeniuk, Wang et Monserrat, « Strong coupling superconductivity in a quasiperiodic host–guest structure », Science Advances, vol. 4, no 4, , eaao4793 (ISSN 2375-2548, PMID 29662950, PMCID 5898833, DOI 10.1126/sciadv.aao4793, lire en ligne)
- Cheng, Y., Wang, J., Rao, T. et He, X., « Pharmaceutical applications of dendrimers: promising nanocarriers for drug discovery », Frontiers in Bioscience, vol. 13, no 13, , p. 1447–1471 (PMID 17981642, DOI 10.2741/2774)
- Cheng, Y. et Xu, T., « Dendrimers as Potential Drug Carriers. Part I. Solubilization of Non-Steroidal Anti-Inflammatory Drugs in the Presence of Polyamidoamine Dendrimers », European Journal of Medicinal Chemistry, vol. 40, no 11, , p. 1188–1192 (PMID 16153746, DOI 10.1016/j.ejmech.2005.06.010)
- Cheng, Y., Xu, T et Fu, R, « Polyamidoamine dendrimers used as solubility enhancers of ketoprofen », European Journal of Medicinal Chemistry, vol. 40, no 12, , p. 1390–1393 (PMID 16226353, DOI 10.1016/j.ejmech.2005.08.002)
- Cheng, Y., Xu, Z, Ma, M. et Xu, T., « Dendrimers as drug carriers: Applications in different routes of drug administration », Journal of Pharmaceutical Sciences, vol. 97, no 1, , p. 123–143 (PMID 17721949, DOI 10.1002/jps.21079)
- D’Emanuele, A et Attwood, D, « Dendrimer–drug interactions », Advanced Drug Delivery Reviews, vol. 57, no 15, , p. 2147–2162 (PMID 16310283, DOI 10.1016/j.addr.2005.09.012)
- Malik, N., Evagorou, E. et Duncan, R., « Dendrimer-platinate: a novel approach to cancer chemotherapy », Anti-Cancer Drugs, vol. 10, no 8, , p. 767–776 (PMID 10573209, DOI 10.1097/00001813-199909000-00010)
- de Silva, A.P., McCaughan, B, McKinney, B.O. F. et Querol, M., « Newer optical-based molecular devices from older coordination chemistry », Dalton Transactions, vol. 10, no 10, , p. 1902–1913 (DOI 10.1039/b212447p)
- Anslyn, E., « Supramolecular Analytical Chemistry », Journal of Organic Chemistry, vol. 72, no 3, , p. 687–699 (PMID 17253783, DOI 10.1021/jo0617971)
- Nguyen, B. et Anslyn, E., « Indicator-displacement assays », Coor. Chem. Rev., vol. 250, nos 23–24, , p. 3118–3127 (DOI 10.1016/j.ccr.2006.04.009)
- Yang, V., Fu, Y., Teng, C. et Ma, S., « A method for the quantitation of protamine in plasma », Thrombosis Research, vol. 74, no 4, , p. 427–434 (PMID 7521974, DOI 10.1016/0049-3848(94)90158-9, lire en ligne)
- Eggleston, A., Rahim, N., Kowalczykowski, S et Ma, S., « A method for the quantitation of protamine in plasma », Nucleic Acids Research, vol. 24, no 7, , p. 1179–1186 (PMID 8614617, PMCID 145774, DOI 10.1093/nar/24.7.1179)
- Biancardi, Tarita, Alberto et Benedetta, « Thiazole orange (TO) as a light-switch probe: a combined quantum-mechanical and spectroscopic study », Physical Chemistry Chemical Physics, vol. 13, no 27, , p. 12595–12602 (PMID 21660321, DOI 10.1039/C1CP20812H)
- Atwood, J, Barbour, L, Jerga, A et Schottel, L, « Guest Transport in a nonporous Organic Solid via Dynamic van der Waals Cooperativity », Science, vol. 298, no 5595, , p. 1000–1002 (PMID 12411698, DOI 10.1126/science.1077591, Bibcode 2002Sci...298.1000A)
- Kitagawa, S et Uemura, K, « Dynamic porous properties of coordination polymers inspired by hydrogen bonds », Chemical Society Reviews, vol. 34, no 2, , p. 109–119 (PMID 15672175, DOI 10.1039/b313997m)
- Sozzani, P, Bracco, S, Commoti, A et Ferretti, R, « Methane and Carbon Dioxide Storage in a Porous van der Waals Crystal », Angewandte Chemie, vol. 44, no 12, , p. 1816–1820 (PMID 15662674, DOI 10.1002/anie.200461704)
- Uemura, K, Kitagawa, S, Fukui, K et Saito, K, « A Contrivance for a Dynamic Porous Framework: Cooperative Guest Adsorption Based on Square Grids Connected by Amide−Amide Hydrogen Bonds », J. Am. Chem. Soc., vol. 126, no 12, , p. 3817–3828 (PMID 15038736, DOI 10.1021/ja039914m)
- Dobrzanska, L, Lloyd, G, Esterhuysen, C et Barbour, L, « Guest-Induced Conformational Switching in a Single Crystal », Angewandte Chemie, vol. 45, no 35, , p. 5856–5859 (PMID 16871642, DOI 10.1002/anie.200602057)
- Eric Hughes, Jason Jordan et Terry Gullion, « Structural Characterization of the [Cs(p-tert-butylcalix[4]arene -H) (MeCN)] Guest–Host System by 13C-133Cs REDOR NMR », Journal of Physical Chemistry B, vol. 105, no 25, , p. 5887–5891 (DOI 10.1021/jp004559x)
- Serkan Erdemir, Mufit Bahadir et Mustafa Yilmaz, « Extraction of Carcinogenic Aromatic Amines from Aqueous Solution Using Calix[n]arene Derivatives as Carriers », Journal of Hazardous Materials, vol. 168, nos 2–3, , p. 1170–1176 (PMID 19345489, DOI 10.1016/j.jhazmat.2009.02.150)