Surface d'énergie potentielle
Une surface d'énergie potentielle est généralement utilisée dans l'approximation adiabatique (ou approximation de Born-Oppenheimer) en mécanique quantique et mécanique statistique afin de modéliser les réactions chimiques et les interactions dans des systèmes chimiques et physiques simples. Le nom de « (hyper)surface » provient du fait que l'énergie totale d'un système atomique peut être représentée comme une courbe ou une surface[note 1], pour laquelle les positions atomiques sont des variables. La meilleure visualisation possible pour cette représentation est de penser à un relief montagneux, pour lequel les directions est-ouest et nord-sud sont deux variables indépendantes (équivalentes par exemple de deux paramètres géométriques de la molécule), et la hauteur du relief l'énergie associée pour des couples de telles variables.
Il existe une correspondance naturelle entre les surfaces d'énergies potentielles comme surfaces polynomiales et leur application dans la théorie du potentiel, qui associe et étudie les fonctions harmoniques en relation avec ces surfaces.
Ainsi, le potentiel de Morse et le puits de potentiel harmonique simple sont des surfaces d'énergies potentielles monodimensionnelles communes (courbes d'énergies potentielles) dans les applications de chimie quantique et de physique quantique.
Ces surfaces d'énergies potentielles (qui peuvent être obtenues analytiquement), cependant, ne donnent de descriptions adéquates que des systèmes chimiques les plus simples. Pour modéliser une réaction chimique donnée, une surface d'énergie potentielle doit être créée pour prendre en compte toutes les orientations possibles[note 2] des molécules de réactant et de produit, et les énergies électroniques de chacune de ces orientations, ce qui s'avère souvent une tâche difficile.
L'énergie électronique est typiquement obtenue pour chacune des dizaines de milliers d'orientations possibles, et ces valeurs d'énergie sont ensuite lissées numériquement sur une fonction multidimensionnelle. La précision de ces points dépendent du niveau de théorie utilisé pour leur calcul. Pour des surfaces particulièrement simples (comme pour la réaction H + H2), la surface de potentiel LEPS (London-Eyring-Polanyi-Sato) dérivée analytiquement peut être suffisante. Les autres méthodes pour obtenir de tels lissages comprennent les cannelures cubiques[note 3], l'interpolation de Shepard, et d'autres types de fonctions de lissage multidimensionnelles.
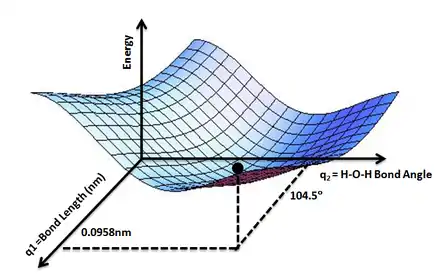
Une fois que la surface d'énergie potentielle a été obtenue, un ensemble de points intéressants doit être déterminé. Le plus important est peut-être le minimum global de l'énergie. Ce minimum global, qui peut être déterminé numériquement, correspond à la configuration nucléaire la plus stable. D'autres points intéressants sont les coordonnées de réaction (le chemin le long duquel sur la surface d'énergie potentielle les atomes « voyagent » durant la réaction chimique), les points selles ou les maxima locaux le long de ces coordonnées (qui correspondent aux états de transition), ainsi que les minima locaux le long de ce chemin (qui correspondent aux intermédiaires de réaction).
Les surfaces d'énergie potentielle sont aussi étudiées par les méthodes expérimentales de la dynamique réactionnelle moléculaire, telles que les faisceaux moléculaires croisés, la chimiluminescence infrarouge et la femtochimie.
Hors la physique et la chimie, les surfaces d'« énergie potentielle » peuvent être associés avec une fonction de coût, qui peut être étudiée afin de minimiser la fonction.
Surfaces attractives et répulsives
Les surfaces d'énergie potentielles des réactions chimiques peuvent être classées comme attractives ou répulsives par comparer les élongations des longueurs des liaisons à l'état de transition par rapport aux réactifs et produits[1],[2]. Pour une réaction du type A + B—C → A—B + C, l'élongation de la liaison A—B nouvellement créée est définie comme R*AB = RAB − R0AB, où RAB est la longueur de liaison A—B à l'état de transition est R0AB à la molécule du produit. De même pour la liaison qui est brisée au cours de la réaction, R*BC = RBC − R0BC, où R0BC réfère à la molécule du réactif.[3]
La surface d'énergie potentielle d'une réaction exothermique est classée comme attractive si R*AB > R*BC, de sorte que l'état de transition est atteinte pendant que les deux réactifs sont encore en approche l'un vers l'autre. Après l'état de transition, la longueur de liaison A—B continue à diminuer, de sorte qu'une grande partie de l'énergie de réaction libérée est transformée en énergie de vibration moléculaire de la liaison A—B[3],[4]. Un exemple serait la réaction harpon K + Br2 → K—Br + Br, à laquelle l'attraction initiale des réactifs à longue distance mène à la formation d'un complexe activé qui ressemble à K+•••Br−•••Br[3]. Les nombres de molécules des produits en excitation vibrationnelle peuvent être mesurés par la chimiluminescence infrarouge[5],[6].
Par contre la surface d'énergie potentielle de la réaction H + Cl2 → HCl + Cl est répulsive parce que R*HCl < R*ClCl et l'état de transition n'est atteint que lors de la séparation des produits[3],[4]. Pour cette réaction où l'atome A (ici H) est plus léger que B ou C, l'énergie de réaction est libérée surtout sous forme d'énergie cinétique translationnelle des produits[3]. Pour une réaction telle que F + H2 → HF + H où l'atome A est plus lourd que B ou C, il y a libération mixte d'énergie, et vibrationnelle et traductionnelle, même si la surface d'énergie potentielle est répulsive[3].
Aux réactions endothermiques, le type de surface détermine le type d'énergie qui induira la réaction de façon la plus efficace. L'énergie translationnelle est la plus efficace pour induire les réactions dont la surface est attractive, tandis que l'excitation est plus efficace lors qu'il s'agit des réactions dôtées d'une surface répulsive[3]. Comme exemple du dernier cas, la réaction F + HCl(v=1)[note 4] → Cl + HF est à peu près cinq fois plus vite que F + HCl(v=0) → Cl + HF, si l'énergie totale du HCl est égale[7].
Histoire
La notion d'une surface d'énergie potentielle est d'abord suggéré par le physicien français René Marcelin en 1913[8]. Le premier calcul semi-empirique d'une surface d'énergie potentielle est effectué par Henry Eyring et Michael Polanyi en 1931. Eyring emploie les surfaces d'énergie potentielle aux calculs des constantes de vitesse par sa théorie de l'état de transition.
Remarque
En raison de la complexité que peuvent atteindre les surfaces d'énergies potentielles de systèmes complexes, d'aucuns, comme Ahmed Zewail, lauréat du prix Nobel de chimie en 1999, préfèrent pour ces cas la dénomination de paysage d'énergie potentielle (en anglais potential energy landscape) et délaissent ainsi la notion de dimension des hypersurfaces.
Notes et références
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Potential energy surface » (voir la liste des auteurs).
Notes
- Parfois multidimensionnelle.
- Autrement dit toutes les coordonnées de réaction possibles.
- En anglais : cubic splines.
- Ici v est le nombre quantique vibrationnel.
Références
- (en) « Attractive potential-energy surface », IUPAC, Compendium of Chemical Terminology [« Gold Book »], Oxford, Blackwell Scientific Publications, 1997, version corrigée en ligne : (2019-), 2e éd. (ISBN 0-9678550-9-8)
- (en) « Repulsive potential-energy surface », IUPAC, Compendium of Chemical Terminology [« Gold Book »], Oxford, Blackwell Scientific Publications, 1997, version corrigée en ligne : (2019-), 2e éd. (ISBN 0-9678550-9-8)
- (en)Laidler, Keith J. Chemical Kinetics (3rd ed., Harper & Row 1987) p. 461-8 (ISBN 0-06-043862-2)
- (en)Steinfeld J.I., Francisco J.S. and Hase W.L. Chemical Kinetics and Dynamics (2nd ed., Prentice-Hall 1998) p. 272-4 (ISBN 0-13-737123-3)
- (en)Steinfeld J.I., Francisco J.S. and Hase W.L. Chemical Kinetics and Dynamics (2nd ed., Prentice-Hall 1998) p. 263 (ISBN 0-13-737123-3)
- Atkins P. et de Paula J. Chimie physique (4e édn. française, de Boeck 2013) p. 851 (ISBN 978-2-8041-6651-9)
- Atkins P. et de Paula J. Chimie physique (4e éd. française, de Boeck 2013) p. 854 (ISBN 978-2-8041-6651-9)
- (en)Computational Chemistry: Introduction to the Theory and Applications of Molecular and Quantum Mechanics Errol G. Lewars, 2nd ed. (Springer 2011) p. 21 (ISBN 978-9048138616)
 Portail de la chimie
Portail de la chimie  Portail de la physique
Portail de la physique