Syndrome d'alpha-tryptasémie héréditaire
Le syndrome d'alpha-tryptasémie héréditaire ou syndrome d'alpha-tryptasémie congénitale ou encore hyper alpha-tryptasémie héréditaire (HαT) (en anglais, hereditary alpha tryptasemia syndrome), est l'expression d'une maladie génétique autosomique dominante, causée par l'augmentation du nombre de copies du gène TPSAB1 (en) codant spécifiquemen l'alpha-tryptase (aussi nommé α-tryptase, tryptase α, ou encore tryptase alpha). Cette maladie a été découverte en 2016, par des chercheurs travaillant au National Institute of Allergy and Infectious Diseases (« institut national des allergies et des maladies infectieuses »). Elle est caractérisée par un taux de tryptase sérique basal élevé et par des symptômes pouvant être associés à un, ou plusieurs systèmes d'organes[1],[2].
| MeSH | C000715748 |
|---|
![]() Mise en garde médicale
Mise en garde médicale
Les porteurs d'une ou de plusieurs copies supplémentaires d'allèles de TPSAB1 codant de l'alpha-tryptase sont appelés porteurs d'alpha-tryptasémie héréditaire. Ils ne développent pas systématiquement ce syndrome, malgré un taux de tryptase sérique basal plus élevé que la moyenne. Les conditions qui expliquent le déclenchement de cette maladie et la variabilité des symptômes n'ont pas encore été identifiées. Il existe un test génétique spécifique qui permet de détecter ces porteurs.
Symptômes
Les symptômes sont hétérogènes et parfois complexes à identifier. Leurs sévérités et leurs variabilités peuvent être très différentes d'un patient à un autre : certains ont peu de symptômes, d'autres sont plus lourdement touchés et ne peuvent pas mener une vie active normale[1],[2],[3].
Les symptômes principaux sont :
- Digestifs
- troubles gastro-intestinaux tels que ballonnements, douleurs abdominales, diarrhées et/ou constipation. Ils sont souvent diagnostiqués comme un syndrome de l'intestin irritable avec des réactions d'intolérances alimentaires et/ou médicamenteuses (49 %),
- troubles stomacaux avec reflux gastriques et/ou brûlures d'estomac (65 %).
- Dermatologiques
- signes d'allergies tels que démangeaisons cutanées ou rougissements associés dans quelques cas à de l'urticaire (51 %),
- réactions allergiques au venin d'insectes hyménoptères tels que guêpes et abeilles (16 %).
- Neurologiques
- symptômes suggérant une dysautonomie : hypotension orthostatique, palpitations, tachycardie, présyncope et syncope, vertiges et/ou difficultés à maintenir un pouls et une pression artérielle normaux. Ces symptômes sont parfois diagnostiqués comme un syndrome de tachycardie orthostatique posturale (= POTS) (46 %).
- Constitutionnels
- douleurs chroniques (musculaires, osseuses) et/ou migraines (47 %),
- arthralgie chronique (45 %),
- sommeil perturbé (39 %).
- Autres
- anomalies du tissu conjonctif, telles que l'hyperlaxité (28 %),
- persistance de la dentition primaire (dents de lait) (21 %),
- anomalie squelettique congénitale (26 %).
La liste de ces symptômes ainsi que les pourcentages cités ont été établis sur une base de données reposant sur 96 sujets réparties dans 35 familles différentes, ayant le syndrome d'alpha-tryptasémie héréditaire
Nombre d'individus porteurs
Le nombre d'individus porteurs d'alpha-tryptasémie héréditaire dans le monde reste encore inconnu ainsi que la proportion finale de personnes développant ce syndrome par rapport au nombre de ces porteurs[1],[4].
Notes importantes : 4 à 6 % de la population générale possède un taux de tryptase sérique basal élevé (>11.4 µg/L), sans que la ou les causes de cette augmentation n'aient été clairement déterminées[3],[5].
Les chercheurs qui ont découvert le syndrome d'alpha-tryptasémie héréditaire ont trouvé que ce taux élevé était dû pour une grande partie des individus, à la présence d'une caractéristique génétique qu'ils ont nommée « alpha-tryptasémie héréditaire ». Ils ont identifié dans une cohorte initiale, 96 sujets de 35 familles présentant un taux de tryptase élevé sérique basale (≥ 9.1 - 39.5 ng/mL) et des caractéristiques cliniques complexes (anomalies familiales du tissu conjonctif dans le contexte de l'atopie et/ou des symptômes souvent associés aux médiateurs des mastocytes, sans mastocytose), qui suivent un modèle d'hérédité autosomique dominante. Des recherches génétiques ont confirmé l'augmentation du nombre de copies du gène TPSAB1 codant l'alpha-tryptase sur un ou deux chromosomes chez tous ces sujets affectés, confirmant par la suite le diagnostic du syndrome de l'alpha-tryptasémie héréditaire[1].
Ces chercheurs ont aussi détecté chez des sujets de deux autres cohortes, des niveaux de tryptase sérique basale élevés (< 8 ng/mL) associés à ces augmentations du nombre de copies du gène TPSAB1 codant l'alpha-tryptase. Parmi ces porteurs d'alpha-tryptasémie héréditaire, les individus affectés ont signalé un ensemble de symptômes semblables à ceux présentés dans la cohorte familiale initiale ci-dessus :
- - L'une de ces 2 cohortes concernait 98 individus répartis dans 53 familles pour lesquels 8 d'entre eux se sont avérés avoir un taux de tryptase sérique basale élevée. Ces 8 individus ont tous été identifiés comme porteurs d'alpha-tryptasémie héréditaire.
- - L'autre cohorte a porté sur 125 individus adultes non apparentés avec 25 d'entre eux retrouvés avec une concentration de tryptase sérique basale élevée. 9 de ces 25 sujets ont été identifiés comme étant porteurs d'alpha-tryptasémie héréditaire (9 autres ont été exclus par manque de couverture génétique) avec 3 d'entre eux atteints du syndrome d'alpha-tryptasémie héréditaire.
Il est à noter que parmi ces 3 cohortes, aucune personne avec un taux de tryptase sérique basal < 8 ng/mL n'a été détectée comme étant porteur d'alpha-tryptasémie héréditaire. Mais des sujets présentant un taux de tryptase sérique basal élevé (pour maximum égal à 9.8 ng/ml) ont toutefois été identifiés comme étant non porteur de cette caractéristique génétique.
Le syndrome d'alpha-tryptasémie héréditaire n'est pas la seule pathologie qui peut expliquer un taux de tryptase sérique élevé. En effet, une élévation temporaire ou chronique de ce taux, peut aussi être due à[6]:
- un syndrome d'activation des mastocytes (= SAMA, ou MCAS en anglais),
- un choc anaphylactique, un œdème ou une allergie grave,
- certains sous-groupes de mastocytoses (cutanées, systémiques),
- plusieurs maladies rares de la moelle osseuse telles que certaines leucémies : leucémie myéloïde aiguë (LMA) ou chronique (LMC), leucémie à éosinophiles chronique (LCE), syndrome myélodysplasique (SMP), néoplasme myéloprolifératif, etc.,
- toute maladie provoquant un trouble d'activation des mastocytes.
Jusqu’à présent, les recherches effectuées ne permettent pas encore de savoir si le syndrome d’alpha-tryptasémie héréditaire est une forme de SAMA, ni s'il peut constituer un sous-groupe de patients atteints de ce syndrome d'activation des mastocytes[3].
Caractéristiques de l'anomalie génétique causant ce syndrome
Le gène TPSAB1 est localisé sur le chromosome 16p13.3 chez l'humain. Il code soit la bêta-tryptase (aussi nommé « β-tryptase », « tryptase β », ou encore « tryptase bêta »), plus précisément la « bêta-1 tryptase » ; soit l'alpha-tryptase, plus précisément « l'alpha-1 tryptase »[7].
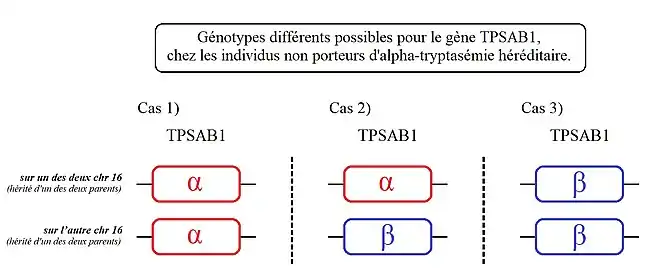
Cas 1) : les personnes présentant ce génotype ont une copie de TPSAB1 codant l'alpha-tryptase sur chacun de leurs chromosomes 16.
Cas 2) : les personnes présentant ce génotype, ont une copie de TPSAB1 codant l'alpha-tryptase sur l'un des deux chromosomes 16 et une autre codant de la bêta-tryptase sur leur autre chromosome 16.
Cas 3) : les personnes présentant ce génotype ont une copie de TPSAB1 codant de la bêta-tryptase sur chacun de leurs chromosomes 16.
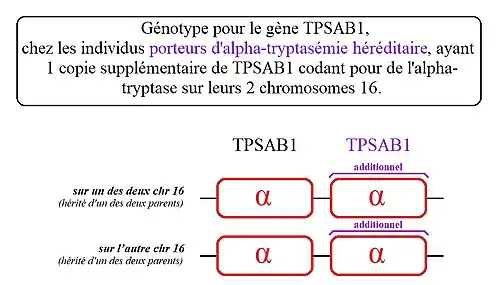
Les individus porteurs d'« alpha-tryptasémie héréditaire » ont une ou plusieurs copies supplémentaires de ce gène codant spécifiquement l'alpha-tryptase sur l'un des deux chromosomes 16, voir plus rarement sur leurs deux chromosomes 16. La conséquence directe est une élévation du taux de tryptase sérique et potentiellement des symptômes associés à ce trouble[1],[2].
Les différents cas jusqu'alors identifiés ayant hérité d'une ou de plusieurs copies supplémentaires de TPSAB1 codant de l'alpha-tryptase :
- 1. Le plus fréquent (un peu moins de 90 % des personnes identifiées concernées), parmi les porteurs d'alpha-tryptasémie héréditaire :
- - des personnes avec une copie supplémentaire de TPSAB1 sur l'un des deux chromosomes 16. Ou parle ici de « duplication» de TPSAB1,
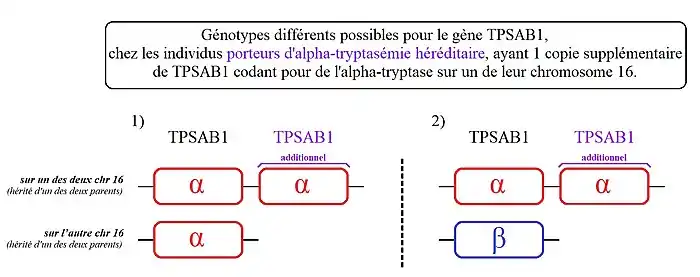
2) Les personnes présentant ce génotype, ont une copie de TPSAB1 codant la bêta-tryptase sur l'un des deux chromosomes 16 et deux copies de TPSAB1 codant l'alpha-tryptase sur leur autre chromosome 16.
- - des personnes avec une copie supplémentaire de TPSAB1 sur l'un des deux chromosomes 16.
- 2. Moins fréquent (moins de 10 % des personnes identifiées concernées, parmi les porteurs d'alpha-tryptasémie héréditaire) :
- Des personnes qui ont deux copies supplémentaires de TPSAB1 sur l'un des deux chromosomes 16. On parle ici de « triplication » du gène TPSAB1.
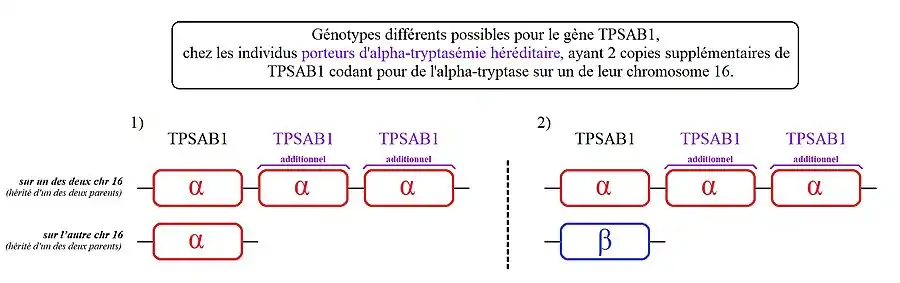
2) Les personnes présentant ce génotype, ont une copie de TPSAB1 codant la bêta-tryptase sur l'un des deux chromosomes 16 et 3 copies de TPSAB1 codant l'alpha-tryptase sur leur autre chromosome 16.
- 3. Plusieurs cas ont été répertoriés avec quatre copies supplémentaires de TPSAB1 sur l'un des deux chromosomes 16. On parle ici de « quintuplation » de TPSAB1[8].
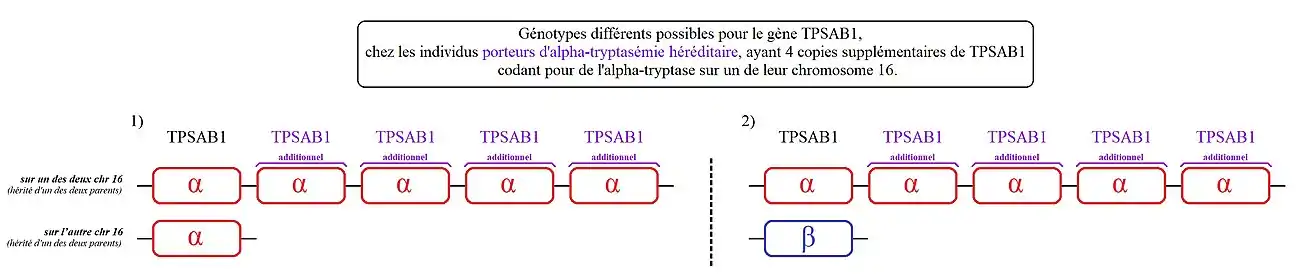
2) Les personnes présentant ce génotype, ont une copie de TPSAB1 codant la bêta-tryptase sur l'un des deux chromosomes 16 et cinq copies de TPSAB1 codant l'alpha-tryptase sur leur autre chromosome 16.
Une corrélation a été observée entre le nombre de ces copies de TPSAB1 supplémentaires, le taux de tryptase basale sérique et la gravité des symptômes des porteurs d'alpha-tryptasémie héréditaire. Plus le nombre de copies de TPSAB1 est élevé, plus le taux de tryptase basale sérique est augmenté et plus la sévérité des symptômes est grande[1],[2]. Un nombre de copies accru de TPSAB1 codant spécifiquement la bêta-tryptase n’a pas été signalé mais n’est pas exclu.
Diagnostic
Le syndrome d'alpha-tryptasémie héréditaire n'a été découvert et caractérisé que très récemment avec la première publication concernant ce syndrome datant de [1]. Seuls quelques spécialistes dans le monde connaissent actuellement ce syndrome.
Le diagnostic se déroule en 3 étapes :
1. Identification des personnes pouvant être atteintes de ce syndrome :
- Toute personne présentant un ou plusieurs des symptômes décrits plus haut pour laquelle aucune cause n'a encore été trouvée, est susceptible de se faire dépister..
- Une attention particulière est accordée aux patients pour lesquels le ou les diagnostics suivants ont déjà été énoncés :
- - syndrome d'activation mastocytaire (= SAMA, ou MCAS en anglais)[3],[9]
- - syndrome de tachycardie posturale (= POTS)[9].
- - syndrome d'Ehlers-Danlos (= SED ou HEDS en anglais) et d'un syndrome d'activation mastocytaire (SAMA), ou dit syndrome d’activation mastocytaire du syndrome d’Ehlers-Danlos (SAMED)[9],[10].
- - syndrome de l'intestin irritable[9].
- La raison principale est que certains couples de symptômes rencontrés dans ces maladies sont parfois très similaires à ceux observés dans le syndrome d'alpha-tryptasemie héréditaire. La confusion entre ce syndrome et ces autres maladies rend le diagnostic souvent très difficile à poser.
2. Dosage du taux de « tryptase sérique »
Le dosage de ce taux se fait par un prélèvement sanguin. Il n'y a aucune mise en condition préalable pour effectuer ce dosage. Cependant, il ne doit pas être fait ni pendant une réaction allergique, ni dans les 10 premiers jours suivants une réaction allergique majeure. Ce type de réaction entraîne en effet une élévation du taux de tryptase sérique, y compris chez les sujets non atteints de ce syndrome, ce qui pourrait donner lieu à une mauvaise interprétation des résultats de ce dosage[11],[12].
- Si le taux de tryptase dans le sang est supérieur à 10 µg/L et si un autre des 2 parents biologiques d'un individu testé a également un taux élevé de tryptase, alors cet individu est beaucoup plus susceptible d'être porteur d'alpha-tryptasémie héréditaire[2],[3].
- Si ce taux se situe entre 7 µg/L et 8 µg/L, il est impossible de confirmer ou infirmer la possibilité que l'individu testé puisse porter une copie surnuméraire du gène TPSAB1 avec ce test seul, bien qu’il soit peu probable que ce soit le cas[3]. Dans cette situation, il est conseillé de recommencer le dosage de la tryptase sérique, tout en dosant si possible ce taux chez les 2 parents de la personne concernée. Il est à noter que des porteurs de ce trait génétique ont déjà été observés avec des taux approchant les 8 µg/L au minimum[1].
- Si le taux de tryptase est inférieur à 7 µg/L, il est très peu probable que l'individu testé soit porteur d'alpha-tryptasémie héréditaire. Il est donc peu probable que les symptômes éprouvés soient dus au syndrome d'alpha-tryptasémie héréditaire[3].
Important : ce syndrome n'est pas la seule cause pouvant expliquer une élévation du taux de tryptase sérique. D'autres pathologies (SAMA, chocs anaphylactiques, allergies graves, mastocytoses, certaines maladies rares de la moelle osseuse[6]...) peuvent provoquer une élévation de ce taux.
D'autres facteurs peuvent aussi faire fluctuer le taux de tryptase sérique. Notamment l'âge :
- - Chez les jeunes enfants, des études faites sur le dosage du taux de tryptase sérique total de nourrissons ont montré que chez les enfants de 0 à 3 mois, les concentrations sériques moyennes de tryptase sont plus élevées que chez les enfants plus âgés et les adultes. Elles diminuent progressivement en atteignant des taux similaires aux jeunes adultes vers 9-12 mois[13],[14]. Plus précisément :
- Une étude portant sur 372 nourrissons de moins de 1 an a révélé une concentration sérique médiane de tryptase de 6,12 ± 3,47 μg/L chez les enfants de 0 à 3 mois, avec une diminution progressive atteignant des concentrations à 3,85 ± 1,8 μg/L vers 9-12 mois[13].
- Dans une autre étude effectuée sur 137 enfants âgés de dix jours à 14 ans, ce sont les nourrissons de moins de 12 mois qui ont présenté les taux de tryptase sérique les plus élevés, avec un maximum entre zéro et trois mois (8,36 ± 2,93 μg/l). Les valeurs étaient stables et basses (entre 3,5 et 4 μg/l) chez les enfants de 12 mois à 14 ans[14].
- - Chez les adultes, une étude portant sur 420 personnes de 18 à 92 ans a montré un taux de tryptase sérique médian de 4.5 μg/L (± 2,1 μg/L). Il s'élève avec l'âge, avec des concentrations qui augmentent en moyenne de manière continue de 0,28 μg/L par décennie[15].
3. Analyse génétique : identification des individus porteurs d'alpha-tryptasémie héréditaire
Il est possible de détecter les individus porteurs d'alpha-tryptasémie héréditaire, avec un test génétique spécifique[2],[3]. Le gène TPSAB1 code soit la bêta-tryptase, soit l'alpha-tryptase. Mais il existe un autre gène qui lui, code exclusivement la bêta-tryptase (plus précisément, la bêta-2 tryptase) : le gène TPSB2 (en), situé juste en amont du gène TPSAB1[16].
Ce test permet d'identifier le nombre total d'allèles codant l'alpha-tryptase ainsi que la bêta-tryptase, afin de pouvoir identifier les porteurs d'alpha-tryptasémie héréditaire. Il se base sur le fait que les individus non porteurs d'alpha-tryptasémie héréditaire, ont 2n copies de TPSB2 codant la bêta-tryptase, et 2n copies de TPSAB1 codant la bêta et/ou alpha-tryptase.

Détails de ce test : Un prélèvement d'échantillon d'ADN des personnes à tester est nécessaire pour ce test (avec par exemple un kit de prélèvement salivaire). Cet ADN est ensuite isolé et amplifié à l'aide d'une technique de réaction en chaîne par polymérase (PCR) en gouttelettes (en) (en anglais : Droplet Digital PCR, ou dit DDPCR).
Limites de ce test : Bien que ce test soit très précis, de rares erreurs de diagnostic peuvent survenir. Ces erreurs peuvent venir :
- d'un mauvais respect du mode d'emploi du kit de prélèvement salivaire, avec notamment :
- - un endommagement de l'échantillon d'ADN des individus à tester, dû à la consommation d'aliments, de boisson, et/ou de tabac 2 heures avant le prélèvement buccal (café, tabac et thé peuvent notamment dégrader cet ADN),
- - d'une contamination de l'échantillon d'ADN testé par la présence d'un ADN extérieur (exemple : proche d'une personne à tester, qui touche par inadvertance la tête de l'écouvillon de cette personne, et qui le contamine avec son propre ADN),
- - d'une confusion dans l'étiquetage des échantillons, notamment lorsque plusieurs personnes à tester sont présentes lors d'un prélèvement d'ADN (risque d'inversion des échantillons envoyés au laboratoire, entre ces personnes).
- D'autres problèmes techniques (par exemple un mauvais conditionnement des échantillons).
- Il a rarement déjà été détecté une variation du nombre de copies codant la bêta-tryptase. Le pourcentage de personnes ayant un gain ou une perte d'allèle codant cette protéine au sein d'une population est inconnu.
- En temps normal, la copie prédictive du nombre d'allèles codant la bêta-tryptase est comprise entre 2 et 4 par individu. C'est-à-dire 2 correspondants aux 2 allèles de TPSB2 et au maximum 2 autres correspondants aux 2 allèles de TPSAB1 (tous situés sur la paire de chromosomes 16).
- Un nombre d'allèles codant la bêta-tryptase inférieur à 2 ou supérieur à 5 peut affecter la capacité de ce test à pouvoir détecter certains porteurs d'alpha-tryptasémie héréditaire.
- Certains génotypes sont impossibles à pouvoir déterminer précisément si d'autres membres de la famille n'ont pas aussi fait ce test. Cela n'altère pas la capacité de cette analyse à pouvoir détecter les porteurs d'alpha-tryptasémie héréditaire en ce cas[2]. Par exemple, pour un individu qui serait porteur de 2 allèles codant la bêta-tryptase et 4 allèles codant l'alpha-tryptase (soit 2β:4α), il sera impossible de déterminer s'il est porteur d'une triplication de TPSAB1 codant l'alpha-tryptase sur un chromosome 16 (avec 1 séquence conservée de TPSAB1 codant l'alpha-tryptase sur son autre chromosome), ou s'il a 2 duplications de TPSAB1 sur ces 2 chromosomes 16.
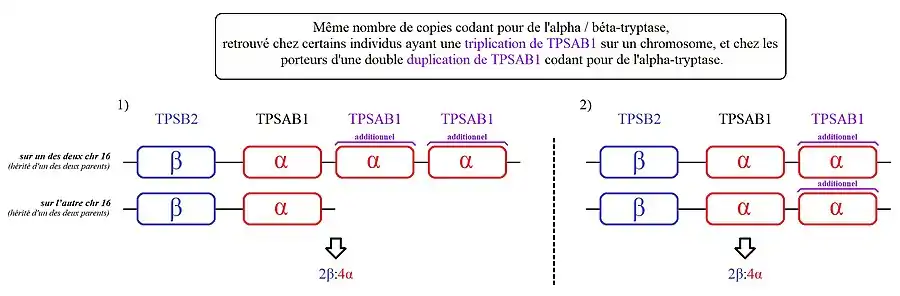
2) Les personnes présentent deux copies codant la bêta-tryptase et quatre copies codant l'alpha-tryptase. Leur génotype est donc aussi 2β:4α.
Actuellement, un seul laboratoire au monde se situant aux États-Unis pratique ce test générique. Celui-ci n'est pas remboursé par la Sécurité sociale française à ce jour. La prescription de ce test doit être délivrée obligatoirement par un médecin avec le consentement du patient, conformément à l’article 16-10 du Code civil[17] :
« L'examen des caractéristiques génétiques d'une personne ne peut être entrepris qu'à des fins médicales ou de recherche scientifique. Le consentement exprès de la personne doit être recueilli par écrit préalablement à la réalisation de l'examen, après qu'elle a été dûment informée de sa nature et de sa finalité. Le consentement mentionne la finalité de l'examen. Il est révocable sans forme et à tout moment. »
Traitements
Il n'existe pas encore de traitements permettant de guérir cette maladie. Les essais cliniques prospectifs manquent pour évaluer l'efficacité des approches de traitement actuelles chez les patients atteints de ce syndrome difficile à traiter. Ces approches thérapeutiques sont actuellement personnalisées pour chaque patient, en fonction de leurs symptômes[3].
Ceux-ci se résument souvent à[18]:
- des traitements antihistaminiques H1 associés aux antihistaminiques H2, pour traiter les symptômes cutanés et gastro-intestinaux, en bloquant certains récepteurs mastocytaires ;
- du cromoglycate de sodium oral si les symptômes gastro-intestinaux sont sévères. Il stabilise les membranes des mastocytes ;
- des auto-injecteurs d'adrénaline (ou d'épinéphrine) pour les personnes présentant des symptômes systémiques graves récurrents et/ou une anaphylaxie. Les déclencheurs de ces symptômes tels que certains aliments, médicaments, piqûres d'insectes, etc. doivent être identifiés et évités.
Risques de transmission de l'alpha-tryptasémie héréditaire
Ce trait génétique se transmet selon le mode autosomique dominant. Cela signifie qu'une personne porteuse d'une seule copie supplémentaire de TPSAB1 sur un de ses 2 chromosomes 16 :
- - a 50 % de probabilité de transmettre ce trait génétique à chacun de ses enfants et cela indépendamment du sexe,
- - a au moins un de ses 2 parents biologiques porteur d'alpha-tryptasémie héréditaire.
C'est pourquoi lorsqu'une personne est diagnostiquée comme en étant porteuse, il est fortement conseillé à sa famille de se faire aussi dépister.
Précisions sur la tryptase et recherches concernant ce syndrome
La tryptase est produite à 99 % par les mastocytes et à moins de 1 % par les basophiles[19],[20]. Chez les mastocytes, elle est secrétée sous 2 formes différentes[21] :
- sous forme immature, avec des monomères d'alpha et/ou bêta-tryptase appelés protryptases. Ces protryptases sont secrétées en continu par les mastocytes et n'ont pas d'activité enzymatique car ce sont des proenzymes[22].
- sous forme mature, adoptant une structure tétramérique faisant partie de la famille des protéases à sérine. Ces tétramères sont produits à partir de protryptases et sont liés à de l'héparine[23]. Ils sont stockés dans les granules de sécrétion des mastocytes[22]. Cette forme de tryptase enzymatiquement active est libérée brutalement vers l'extérieur des mastocytes en cas de stimuli spécifiques, accompagnée d'autres médiateurs préformés vasoactifs (en), pro-inflammatoires et nociceptifs. Ce phénomène de libération est appelé : « dégranulation mastocytaire ».
La tryptase peut cliver plusieurs protéines. Certains de ses substrats ont déjà été identifiés et testés in vitro : la tryptase clive et inactive le fibrinogène[24],[25],[26], active la métalloprotéase 3 promatricielle (proMMP-3)[27]. La tryptase dégrade la fibronectine[28], active la pro-urokinase[29] et génère C3a à partir du complément C3[30]. Le peptide intestinal vasoactif (VIP) (ex vivo et in vitro)[31],[32] et le peptide associé au gène de la calcitonine[33] sont également dégradés par la tryptase. Elle stimule la prolifération des cellules musculaires lisses, des fibroblastes et des cellules épithéliales[34],[35],[36] et la synthèse du collagène de type I par les fibroblastes humains[37],[38],[39]. Elle clive le kininogène de bas et haut poids moléculaire[40],[41],[42] ce qui augmente la perméabilité vasculaire par activation de la prékallikréine et par la production directe de bradykinine[43]. La tryptase peut théoriquement jouer un rôle dans l'athérosclérose en dégradant les lipoprotéines de haute densité (HDL) et en empêchant ainsi l'élimination du cholestérol par celles-ci[44].
- 3 structures tétramériques différentes de cette forme mature de tryptase ont été décrites, à savoir :
- des homotétramères composés de 4 protomères de bêta-tryptase, appelés bêta-tryptase.
- Aucun sujet dépourvu de gène codant une forme active de bêta-tryptase n'a encore été rapporté[45].
- Des homotétramères composés de 4 protomères d'alpha-tryptase, appelés alpha-tryptase (observés in vitro seulement) qui présentent une activité protéolytique négligeable[46].
- Les protryptases sont secrétées continuellement par les mastocytes[22]. Mais seuls les monomères d'alpha-tryptase semblent exclusivement associés à cette voix de sécrétion[47], les monomères de bêta-tryptase pouvant être dirigés vers les granules de sécrétion des mastocytes pour devenir des tétramères. L'alpha-tryptase sous forme de tétramère n'a pu être observée et étudiée qu'in vivo[48]. Ces homotétramères matures d'alpha-tryptase présentent une activité protéolytique négligeable, expliquée en grande partie par la présence d'un acide aspartique au lieu d'une glycine dans l'une des boucles qui forment sa fente de liaison au substrat. Ce qui empêche ces substrats potentiels d'atteindre le site catalytique des homotétramères d'alpha-tryptase et d'être dégradés[46],[49].
- Note : une étude a rapporté que sur 274 personnes étudiées, 30 % en moyenne portent des allèles de TPSAB1 codant uniquement la bêta-tryptase et ne produisent pas d'alpha-tryptase. Ce déficit est courant et varie considérablement entre les groupes ethniques. Aucun phénotype clinique dû à ce manque n'a encore été observé[50].
- Des hétérotétramères composés de 2 protomères d'alpha-tryptase, et de 2 protomères de bêta-tryptase, appelés alpha/bêta-tryptase (nommé aussi α/β-tryptase, observées in vivo et in vitro).
- Une étude publiée en a montré que les alpha/bêta-tryptases se forment naturellement dans les mastocytes des individus exprimant l'alpha-tryptase et donc porteurs d'au moins un allèle TPSAB1 codant cette protéine[51]. Ces hétérotétramères d'alpha/bêta-tryptases représentent une part importante de l’activité enzymatique totale de la tryptase dont la presque l'entièreté est stockée dans des granules de sécrétion mastocytaires. Cette étude suggère que les monomères individuels d'alpha et bêta-tryptases ne se combinent pas de manière aléatoire mais se forment plutôt à partir d'homodimères d'alpha et bêta-tryptases, dont l'existence a été prédite sur la base de la cinétique de formation du monomère en tétramère[52]. Elle a aussi mis en évidence que l'alpha/bêta-tryptase, contrairement à la bêta-tryptase
- → active le récepteur PAR2 in vitro.
- D'autres études ont montré que ce récepteur est exprimé sur des types de cellules telles que des cellules du muscle lisse, du système nerveux périphérique et central, de l'endothélium et est hautement exprimé dans les poumons, le foie, le tube digestif, la peau et les vaisseaux sanguins[53]. PAR2 joue un rôle important dans l'inflammation, la nociception avec l'induction et le maintien de la douleur persistante, en particulier dans les douleurs inflammatoires, neuropathiques, cancéreuses[54],[55] et contribue à l'obésité, ainsi qu'au dysfonctionnement métabolique[56].
- → facilite la dégranulation des mastocytes in vivo et in vitro, lorsque la peau est soumise à des vibrations.
- L'alpha/bêta-tryptase clive alors la sous-unité α des récepteurs transmembranaires EMR2 (EGF-like module-containing mucin-like hormone receptor-like 2), situés à la surface des mastocytes. Les chercheurs de cette même étude pensent que ce clivage par l'alpha/bêta-tryptase affaiblit probablement l’association de cette sous-unité α avec le sulfate de dermatane contenu dans le tissu conjonctif de la peau. Cela engendre la séparation entre cette même sous-unité α et la sous-unité β auparavant liées de ces récepteurs EMR2. Une cascade de réactions biochimiques à l'intérieur des mastocytes aboutissant à la dégranulation de ces cellules et à des réactions urticaires s'ensuit. Ces chercheurs ont émis comme hypothèse que chez les personnes atteintes d'alpha-tryptasemie héréditaire, l'alpha/bêta-tryptase contribue probablement à ces réactions urticaires vibratoires provoquées chez presque tous les patients touchés par ces symptômes.
- Ainsi, l’activation de PAR2 et/ou EMR2 par l'alpha/bêta-tryptase peut contribuer au prurit, à la dysautonomie et aux symptômes de douleur associés à l'alpha-tryptasémie héréditaire.
- L'existence d'autres différences dans la spécificité de substrats entre la bêta-tryptase et l'alpha/bêta-tryptase restent à présager. Ce nouveau répertoire de substrats pourrait aussi être la cause de certaines des caractéristiques cliniques de l'alpha-tryptasémie héréditaire ainsi que d'autres troubles impliquant les mastocytes.
- Ces différences de substrats sont plausiblement explicables par des effets allostériques différents des protomères d'alpha-tryptase sur les protomères de bêta-tryptase voisins dans les molécules d'alpha/bêta-tryptase, par rapport aux effets allostériques des protomères de bêta-tryptase sur les autres protomères voisins[57] dans les molécules de bêta-tryptase.
- D'une manière générale, toujours selon cette étude, l'alpha/bêta-tryptase devrait être considérée comme un médiateur pouvant influencer la gravité des troubles induits par les mastocytes au niveau des tissus où ils sont naturellement abondants, comme dans les tissus muqueux, le derme et les sites périvasculaires, ou lorsqu’ils s’accumulent dans le cadre d’un processus pathologique, tel que dans le muscle lisse bronchique des personnes asthmatiques[58]..
À noter que la méthode utilisée pour mesurer la « tryptase sérique totale » (ImmunoCAP) permet de doser sans les distinguer, les formes de tryptases matures et immatures contenues dans le sérum[12].
_VF.jpg.webp)
D'autres pistes on déjà été explorées : Le locus contenant le gène de CACNA1H (en) est adjacent au locus de la tryptase (ce dernier comportant 5 gènes : TPSG1 (en), TPSB2, TPSAB1, et TPSD1 (en)). CACNA1H code la sous-unité α1H nommée Cav3.2, du canal calcique « voltage dépendant » de type T. Les canaux calciques de type T dépendant de la tension basse sont exprimés dans l'ensemble du système nerveux, où ils jouent un rôle essentiel dans la formation de l'excitabilité neuronale. Il contribue notamment à la transmission de signaux nociceptifs[59],[60]. Actuellement, on en sait peu sur les mécanismes cellulaires contrôlant l'expression et la fonction des canaux de type T.
- - Un haplotype a été identifié comme étant couramment cohérité avec des duplications de TPSAB1, chez environ 2 tiers des individus testés ayant le syndrome d'alpha tryptasémie héréditaire[61]. Cet haplotype comprend trois variations génétiques (induites respectivement par trois mutations faux-sens) présentes sur un même allèle de CACNA1H.
- Aucun effet clinique induit par ces variations n'a été détecté à l'état hétérozygote, malgré la constatation in vitro d'un gain partiel de fonction des canaux calciques Cav3.2 codés par cet allèle de CACNA1H portant ces 3 variations génétiques. L'importance de ces variations sur les phénotypes cliniques potentiels à l'état homozygote n'a pas encore été explorée. Aucune étude sur leurs effets éventuels au-delà des symptômes associés au nombre accru de copies de TPSAB1 n'a encore été effectuée.
- - 2 variations génétiques présentes sur un l'allèle de TPSG1 (induits respectivement par 2 mutations faux-sens), codant la gamma-tryptase (en) étaient également présentes exclusivement et fréquemment rencontrées avec l'haplotype décrit ci-dessus, dans les familles testées. Les effets potentiels de ces variations n’ont pas encore été étudiés in vitro.
Quelques précisions : la gamma-tryptase (aussi nommée « γ-tryptase », « tryptase γ », « tryptase gamma », ou encore « Tryptase transmembranaire (TMT) ») codée par TPSG1 est une protéase à sérine avec a une structure assez semblable à celle des alpha- et bêta-tryptases. Avec notamment 50 % de la séquence d'acides aminés de son domaine catalytique semblable à celle de la bêta-1 tryptase (codé par TPSAB1). Malgré certaines similitudes structurelles, la gamma-tryptase semble avoir des fonctionnalités différentes avec certaines activités protéolytiques distinctes de la bêta 1 tryptase. La gamma-tryptase n’est pas connu pour être sécrétée. Elle est seulement stockée dans les granules de sécrétion et est exprimée de manière transitoire sur la surface externe de la membrane plasmique des mastocytes grâce à son domaine transmembranaire après leur dégranulation.
Des expériences ont montré que la gamma-tryptase à la capacité d'induire une hyperréactivité bronchique dans les poumons des rats en activant une voie dépendante des interleukines IL-13, IL-4Rα, et STAT6[62]. Mais de nombreux travaux supplémentaires sont nécessaires pour comprendre le rôle fonctionnel de la gamma-tryptase de type sauvage dans les mastocytes humains avant de pouvoir entreprendre l'analyse fonctionnelle de l'allèle de TPSG1 portant ces 2 variations génétiques.
En conclusion, le lien entre l'augmentation du nombre de copies du gène TPSAB1 codant l'alpha-tryptase et les symptômes éprouvés par les personnes atteintes d'alpha-tryptasémie héréditaire n'a pas encore été clairement défini. Ce domaine reste un sujet nécessitant des recherches spécifiques, pour être mieux compris et approfondi.
Notes et références
- Jonathan J. Lyons, Xiaomin Yu, Jason D. Hughes et Quang T. Le, « Elevated basal serum tryptase identifies a multisystem disorder associated with increased TPSAB1 copy number », Nature Genetics, vol. 48, no 12, , p. 1564–1569 (ISSN 1546-1718, PMID 27749843, PMCID 5397297, DOI 10.1038/ng.3696, lire en ligne, consulté le )
- Jonathan J. Lyons, « Hereditary alpha tryptasemia: genotyping and associated clinical features », Immunology and allergy clinics of North America, vol. 38, no 3, , p. 483–495 (ISSN 0889-8561, PMID 30007465, PMCID 6411063, DOI 10.1016/j.iac.2018.04.003, lire en ligne, consulté le )
- « Hereditary Alpha Tryptasemia and Hereditary Alpha Tryptasemia Syndrome FAQ | NIH: National Institute of Allergy and Infectious Diseases », sur www.niaid.nih.gov (consulté le )
- Quang T. Le, Jonathan J. Lyons, Andrea N. Naranjo et Ana Olivera, « Impact of naturally forming human α/β-tryptase heterotetramers in the pathogenesis of hereditary α-tryptasemia », The Journal of Experimental Medicine, (ISSN 1540-9538, PMID 31337736, DOI 10.1084/jem.20190701, lire en ligne, consulté le )
- C. Fellinger, W. Hemmer, S. Wöhrl et G. Sesztak-Greinecker, « Clinical characteristics and risk profile of patients with elevated baseline serum tryptase », Allergologia Et Immunopathologia, vol. 42, no 6, , p. 544–552 (ISSN 1578-1267, PMID 25224360, DOI 10.1016/j.aller.2014.05.002, lire en ligne, consulté le )
- Peter Valent, Wolfgang R. Sperr, Karl Sotlar et Andreas Reiter, « The serum tryptase test: an emerging robust biomarker in clinical hematology », Expert Review of Hematology, vol. 7, no 5, , p. 683–690 (ISSN 1747-4094, PMID 25169217, PMCID 4603354, DOI 10.1586/17474086.2014.955008, lire en ligne, consulté le )
- « TPSAB1 tryptase alpha/beta 1 [Homo sapiens (human)] - Gene - NCBI », sur www.ncbi.nlm.nih.gov (consulté le )
- Vito Sabato, Jack Chovanec, Margaretha Faber et Joshua D. Milner, « First Identification of an Inherited TPSAB1 Quintuplication in a Patient with Clonal Mast Cell Disease », Journal of Clinical Immunology, vol. 38, no 4, , p. 457–459 (ISSN 1573-2592, PMID 29748908, DOI 10.1007/s10875-018-0506-y, lire en ligne, consulté le )
- « Hereditary alpha tryptasemia syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program », sur rarediseases.info.nih.gov (consulté le )
- « "SED ou SAMA SAMA et SED SAMED" par le Dr D. GROSSIN, Président du Groupe d’Etude et de Recherche (= GERSED) de France », sur http://www.gersed.com/ (consulté le )
- « Labtest - Tryptase », sur www.labtestsonline.fr (consulté le )
- « ImmunoCAP Tryptase - Phadia - Setting the Standard - Phadia.com », sur www.phadia.com (consulté le )
- Wahib Belhocine, Zouher Ibrahim, Véronique Grandné et Christophe Buffat, « Total serum tryptase levels are higher in young infants », Pediatric Allergy and Immunology: Official Publication of the European Society of Pediatric Allergy and Immunology, vol. 22, no 6, , p. 600–607 (ISSN 1399-3038, PMID 21736626, DOI 10.1111/j.1399-3038.2011.01166.x, lire en ligne, consulté le )
- Z. Ibrahim, P. Bongrand, P. Carayon et J. Vitte, « Détermination du taux sérique de tryptase dans une population pédiatrique », Revue Française d'Allergologie, vol. 49, no 7, , p. 524–527 (ISSN 1877-0320, DOI 10.1016/j.reval.2009.05.001, lire en ligne, consulté le )
- Arturo Gonzalez-Quintela, Luis Vizcaino, Francisco Gude et Jesus Rey, « Factors influencing serum total tryptase concentrations in a general adult population », Clinical Chemistry and Laboratory Medicine, vol. 48, no 5, , p. 701–706 (ISSN 1437-4331, PMID 20178448, DOI 10.1515/CCLM.2010.124, lire en ligne, consulté le )
- « TPSB2 tryptase beta 2 (gene/pseudogene) [Homo sapiens (human)] - Gene - NCBI », sur www.ncbi.nlm.nih.gov (consulté le )
- Code civil - Article 16-10 (lire en ligne)
- (en) Jonathan J. Lyons, « Hereditary Alpha Tryptasemia », Immunology and Allergy Clinics of North America, vol. 38, no 3, , p. 483–495 (DOI 10.1016/j.iac.2018.04.003, lire en ligne, consulté le )
- Sherryline Jogie-Brahim, Hae-Ki Min, Yoshihiro Fukuoka et Han-Zhang Xia, « Expression of α-tryptase and β-tryptase by human basophils », Journal of Allergy and Clinical Immunology, vol. 113, no 6, , p. 1086–1092 (ISSN 0091-6749, DOI 10.1016/j.jaci.2004.02.032, lire en ligne, consulté le )
- (en) Barbara Foster, Lawrence B. Schwartz, Gilles Devouassoux et Dean D. Metcalfe, « Characterization of mast-cell tryptase-expressing peripheral blood cells as basophils », Journal of Allergy and Clinical Immunology, vol. 109, no 2, , p. 287–293 (DOI 10.1067/mai.2002.121454, lire en ligne, consulté le )
- Joana Vitte, « Human mast cell tryptase in biology and medicine », Molecular Immunology, vol. 63, no 1, , p. 18–24 (ISSN 0161-5890, DOI 10.1016/j.molimm.2014.04.001, lire en ligne, consulté le )
- (en) Yoshihiro Fukuoka, George Moxley, Wei Zhao et Jiang Hu, « Tryptase Precursors Are Preferentially and Spontaneously Released, Whereas Mature Tryptase Is Retained by HMC-1 Cells, Mono-Mac-6 Cells, and Human Skin-Derived Mast Cells », The Journal of Immunology, vol. 170, no 11, , p. 5667–5673 (ISSN 0022-1767 et 1550-6606, PMID 12759448, DOI 10.4049/jimmunol.170.11.5667, lire en ligne, consulté le )
- K. Sakai, S. Ren et L. B. Schwartz, « A novel heparin-dependent processing pathway for human tryptase. Autocatalysis followed by activation with dipeptidyl peptidase I », The Journal of Clinical Investigation, vol. 97, no 4, , p. 988–995 (ISSN 0021-9738, PMID 8613553, DOI 10.1172/JCI118523, lire en ligne, consulté le )
- (en) L. B. Schwartz, T. R. Bradford, B. H. Littman et B. U. Wintroub, « The fibrinogenolytic activity of purified tryptase from human lung mast cells. », The Journal of Immunology, vol. 135, no 4, , p. 2762–2767 (ISSN 0022-1767 et 1550-6606, PMID 3161948, lire en ligne, consulté le )
- (en) L. B. Schwartz, C. M. Baumgarten, M. Carr et A. E. Lawson, « Human tryptase fibrinogenolysis is optimal at acidic pH and generates anticoagulant fragments in the presence of the anti-tryptase monoclonal antibody B12. », The Journal of Immunology, vol. 159, no 7, , p. 3540–3548 (ISSN 0022-1767 et 1550-6606, PMID 9317153, lire en ligne, consulté le )
- Vanessa A. Thomas, Christine J. Wheeless, M. Sharon Stack et David A. Johnson, « Human Mast Cell Tryptase Fibrinogenolysis: Kinetics, Anticoagulation Mechanism, and Cell Adhesion Disruption† », Biochemistry, vol. 37, no 8, , p. 2291–2298 (ISSN 0006-2960 et 1520-4995, DOI 10.1021/bi972119z, lire en ligne, consulté le )
- B L Gruber, M J Marchese, K Suzuki et L B Schwartz, « Synovial procollagenase activation by human mast cell tryptase dependence upon matrix metalloproteinase 3 activation. », Journal of Clinical Investigation, vol. 84, no 5, , p. 1657–1662 (ISSN 0021-9738, DOI 10.1172/jci114344, lire en ligne, consulté le )
- Jouko Lohi, Jorma Kesksi-Oja et Ilkka Harvima, « Pericellulars substrates of human mast cell tryptase: 72,000 Dalton gelatinase and fibronectin », Journal of Cellular Biochemistry, vol. 50, no 4, , p. 337–349 (ISSN 0730-2312 et 1097-4644, DOI 10.1002/jcb.240500402, lire en ligne, consulté le )
- M. Sharon Stack et David A. Johnson, « Single chain urinary-type plasminogen activator (prourokinase) is activated by human mast cell tryptase », Fibrinolysis, vol. 8, , p. 34 (ISSN 0268-9499, DOI 10.1016/0268-9499(94)90379-4, lire en ligne, consulté le )
- L. B. Schwartz, M. S. Kawahara, T. E. Hugli et D. Vik, « Generation of C3a anaphylatoxin from human C3 by human mast cell tryptase », Journal of Immunology (Baltimore, Md.: 1950), vol. 130, no 4, , p. 1891–1895 (ISSN 0022-1767, PMID 6339618, lire en ligne, consulté le )
- E. K. Tam et G. H. Caughey, « Degradation of airway neuropeptides by human lung tryptase », American Journal of Respiratory Cell and Molecular Biology, vol. 3, no 1, , p. 27–32 (ISSN 1044-1549, PMID 1694672, DOI 10.1165/ajrcmb/3.1.27, lire en ligne, consulté le )
- G. M. Franconi, P. D. Graf, S. C. Lazarus et J. A. Nadel, « Mast cell tryptase and chymase reverse airway smooth muscle relaxation induced by vasoactive intestinal peptide in the ferret », The Journal of Pharmacology and Experimental Therapeutics, vol. 248, no 3, , p. 947–951 (ISSN 0022-3565, PMID 2495355, lire en ligne, consulté le )
- Andrew F. Walls, Susan D. Brain, Anita Desai et Peter J. Jose, « Human mast cell tryptase attenuates the vasodilator activity of calcitonin generelated peptide », Biochemical Pharmacology, vol. 43, no 6, , p. 1243–1248 (ISSN 0006-2952, DOI 10.1016/0006-2952(92)90498-8, lire en ligne, consulté le )
- T. Hartmann, S. J. Ruoss, W. W. Raymond et K. Seuwen, « Human tryptase as a potent, cell-specific mitogen: role of signaling pathways in synergistic responses », American Journal of Physiology-Lung Cellular and Molecular Physiology, vol. 262, no 5, , L528–L534 (ISSN 1040-0605 et 1522-1504, DOI 10.1152/ajplung.1992.262.5.l528, lire en ligne, consulté le )
- J. A. Cairns et A. F. Walls, « Mast cell tryptase is a mitogen for epithelial cells. Stimulation of IL-8 production and intercellular adhesion molecule-1 expression », Journal of Immunology (Baltimore, Md.: 1950), vol. 156, no 1, , p. 275–283 (ISSN 0022-1767, PMID 8598474, lire en ligne, consulté le )
- J. K. Brown, C. L. Tyler, C. A. Jones et S. J. Ruoss, « Tryptase, the dominant secretory granular protein in human mast cells, is a potent mitogen for cultured dog tracheal smooth muscle cells », American Journal of Respiratory Cell and Molecular Biology, vol. 13, no 2, , p. 227–236 (ISSN 1044-1549, PMID 7626290, DOI 10.1165/ajrcmb.13.2.7626290, lire en ligne, consulté le )
- J A Cairns et A F Walls, « Mast cell tryptase stimulates the synthesis of type I collagen in human lung fibroblasts. », Journal of Clinical Investigation, vol. 99, no 6, , p. 1313–1321 (ISSN 0021-9738, DOI 10.1172/jci119290, lire en ligne, consulté le )
- J. Zhang, B. L. Gruber, M. J. Marchese et S. Zucker, « Mast cell tryptase does not alter matrix metalloproteinase expression in human dermal fibroblasts: further evidence that proteolytically-active tryptase is a potent fibrogenic factor », Journal of Cellular Physiology, vol. 181, no 2, , p. 312–318 (ISSN 0021-9541, PMID 10497310, DOI 10.1002/(SICI)1097-4652(199911)181:23.0.CO;2-1, lire en ligne, consulté le )
- B. L. Gruber, R. R. Kew, A. Jelaska et M. J. Marchese, « Human mast cells activate fibroblasts: tryptase is a fibrogenic factor stimulating collagen messenger ribonucleic acid synthesis and fibroblast chemotaxis », Journal of Immunology (Baltimore, Md.: 1950), vol. 158, no 5, , p. 2310–2317 (ISSN 0022-1767, PMID 9036979, lire en ligne, consulté le )
- Lawrence B. Schwartz, Manfred Maier et Jocelyn Spragg, « Interaction of Human Low Molecular Weight Kininogen with Human Mast Cell Tryptase », dans Kinins IV, Springer US, (ISBN 9781468451450, lire en ligne), p. 105–111
- David Proud, Edward S. Siekierski et Graham S. Bailey, « Identification of human lung mast cell kininogenase as tryptase and relevance of tryptase kininogenase activity », Biochemical Pharmacology, vol. 37, no 8, , p. 1473–1480 (ISSN 0006-2952, DOI 10.1016/0006-2952(88)90008-1, lire en ligne, consulté le )
- ANDREW F. WALLS, AMANDA R. BENNETT, JAVIER SUEIRAS-DIAZ et HÅKAN OLSSON, « The kininogenase activity of human mast cell tryptase », Biochemical Society Transactions, vol. 20, no 3, , p. 260S–260S (ISSN 0300-5127 et 1470-8752, DOI 10.1042/bst020260s, lire en ligne, consulté le )
- T. Imamura, A. Dubin, W. Moore et R. Tanaka, « Induction of vascular permeability enhancement by human tryptase: dependence on activation of prekallikrein and direct release of bradykinin from kininogens », Laboratory Investigation; a Journal of Technical Methods and Pathology, vol. 74, no 5, , p. 861–870 (ISSN 0023-6837, PMID 8642782, lire en ligne, consulté le )
- Lee Miriam, Sommerhoff Christian P., von Eckardstein Arnold et Zettl Frank, « Mast Cell Tryptase Degrades HDL and Blocks Its Function as an Acceptor of Cellular Cholesterol », Arteriosclerosis, Thrombosis, and Vascular Biology, vol. 22, no 12, , p. 2086–2091 (DOI 10.1161/01.ATV.0000041405.07367.B5, lire en ligne, consulté le )
- Richard L Stevens, « Faculty of 1000 evaluation for Human subjects are protected from mast cell tryptase deficiency despite frequent inheritance of loss-of-function mutations. », sur F1000 - Post-publication peer review of the biomedical literature, (consulté le )
- Chifu Huang, Lixin Li, Steven A. Krilis et Kara Chanasyk, « Human Tryptases α and β/II Are Functionally Distinct Due, in Part, to a Single Amino Acid Difference in One of the Surface Loops That Forms the Substrate-binding Cleft », Journal of Biological Chemistry, vol. 274, no 28, , p. 19670–19676 (ISSN 0021-9258 et 1083-351X, DOI 10.1074/jbc.274.28.19670, lire en ligne, consulté le )
- « A novel heparin-dependent processing pathway for human tryptase. Autocatalysis followed by activation with dipeptidyl peptidase I. », Journal of Clinical Investigation, vol. 97, no 4, , p. 988–995 (ISSN 0021-9738, PMID 8613553, lire en ligne, consulté le )
- Trevor Selwood, Zhi-Mei Wang, Darrell R. McCaslin et Norman M. Schechter, « Diverse stability and catalytic properties of human tryptase alpha and beta isoforms are mediated by residue differences at the S1 pocket », Biochemistry, vol. 41, no 10, , p. 3329–3340 (ISSN 0006-2960, PMID 11876641, DOI 10.1021/bi015662v, lire en ligne, consulté le )
- Ulf Marquardt, Frank Zettl, Robert Huber et Wolfram Bode, « The crystal structure of human alpha1-tryptase reveals a blocked substrate-binding region », Journal of Molecular Biology, vol. 321, no 3, , p. 491–502 (ISSN 0022-2836, PMID 12162961, DOI 10.1016/s0022-2836(02)00625-3, lire en ligne, consulté le )
- D. Soto, C. Malmsten, J. L. Blount et D. J. Muilenburg, « Genetic deficiency of human mast cell alpha-tryptase », Clinical and Experimental Allergy: Journal of the British Society for Allergy and Clinical Immunology, vol. 32, no 7, , p. 1000–1006 (ISSN 0954-7894, PMID 12100045, DOI 10.1046/j.1365-2222.2002.01416.x, lire en ligne, consulté le )
- (en) « Impact of naturally forming human α/β-tryptase heterotetramers in the pathogenesis of hereditary α-tryptasemia | Request PDF », sur ResearchGate (consulté le )
- (en) Shunlin Ren, Kentaro Sakai et Lawrence B. Schwartz, « Regulation of Human Mast Cell β-Tryptase: Conversion of Inactive Monomer to Active Tetramer at Acid pH », The Journal of Immunology, vol. 160, no 9, , p. 4561–4569 (ISSN 0022-1767 et 1550-6606, PMID 9574563, lire en ligne, consulté le )
- « F2RL1 F2R like trypsin receptor 1 [Homo sapiens (human)] - Gene - NCBI », sur www.ncbi.nlm.nih.gov (consulté le )
- G. S. Cottrell, S. Amadesi, F. Schmidlin et N. Bunnett, « Protease-activated receptor 2: activation, signalling and function », Biochemical Society Transactions, vol. 31, no Pt 6, , p. 1191–1197 (ISSN 0300-5127, PMID 14641024, DOI 10.1042/bst0311191, lire en ligne, consulté le )
- P. Mrozkova, J. Palecek et D. Spicarova, « The role of protease-activated receptor type 2 in nociceptive signaling and pain », Physiological Research, vol. 65, no 3, , p. 357–367 (ISSN 1802-9973, PMID 27070742, lire en ligne, consulté le )
- Junxian Lim, Abishek Iyer, Ligong Liu et Jacky Y. Suen, « Diet-induced obesity, adipose inflammation, and metabolic dysfunction correlating with PAR2 expression are attenuated by PAR2 antagonism », The FASEB Journal, vol. 27, no 12, , p. 4757–4767 (ISSN 0892-6638 et 1530-6860, DOI 10.1096/fj.13-232702, lire en ligne, consulté le )
- Henry R. Maun, Peter S. Liu, Yvonne Franke et Charles Eigenbrot, « Dual functionality of β-tryptase protomers as both proteases and cofactors in the active tetramer », The Journal of Biological Chemistry, vol. 293, no 25, 06 22, 2018, p. 9614–9628 (ISSN 1083-351X, PMID 29661938, PMCID 6016454, DOI 10.1074/jbc.M117.812016, lire en ligne, consulté le )
- Christopher E. Brightling, Peter Bradding, Fiona A. Symon et Stephen T. Holgate, « Mast-cell infiltration of airway smooth muscle in asthma », The New England Journal of Medicine, vol. 346, no 22, , p. 1699–1705 (ISSN 1533-4406, PMID 12037149, DOI 10.1056/NEJMoa012705, lire en ligne, consulté le )
- Agustin García-Caballero, Vinicius M. Gadotti, Patrick Stemkowski et Norbert Weiss, « The Deubiquitinating Enzyme USP5 Modulates Neuropathic and Inflammatory Pain by Enhancing Cav3.2 Channel Activity », Neuron, vol. 83, no 5, , p. 1144–1158 (ISSN 0896-6273, DOI 10.1016/j.neuron.2014.07.036, lire en ligne, consulté le )
- S. Choi, H. S. Na, J. Kim et J. Lee, « Attenuated pain responses in mice lacking CaV3.2 T-type channels », Genes, Brain and Behavior, vol. 6, no 5, , p. 425–431 (ISSN 1601-1848 et 1601-183X, DOI 10.1111/j.1601-183x.2006.00268.x, lire en ligne, consulté le )
- (en) Joshua D. Milner, Michael A. Colicos, Leslie G. Biesecker et Heejong Sung, « A common haplotype containing functional CACNA1H variants is frequently coinherited with increased TPSAB1 copy number », Genetics in Medicine, vol. 20, no 5, , p. 503–512 (ISSN 1530-0366, DOI 10.1038/gim.2017.136, lire en ligne, consulté le )
- (en) « (PDF) Biochemical and Functional Characterization of Human Transmembrane Tryptase (TMT)/Tryptase γ », sur ResearchGate (consulté le )
 Portail de la médecine
Portail de la médecine