Tumeur fibreuse solitaire
Une tumeur fibreuse solitaire est une tumeur rare le plus souvent bénigne, parfois très volumineuse, développée à partir des fibroblastes contenus dans les tissus mous de l'organisme. La plèvre, dans le thorax, est le site le plus fréquent, mais toute région anatomique est susceptible de développer l'une de ces tumeurs. La première description remonte à 1931.
| Spécialité | Oncologie |
|---|
| ICD-O | M8815/0 |
|---|---|
| MedlinePlus | 000116 |
| MeSH | D054364 |
![]() Mise en garde médicale
Mise en garde médicale
Les signes cliniques sont souvent frustes, en raison de la lenteur d'évolution ; ils apparaissent lorsque la tumeur, qui peut être très volumineuse, comprime les organes voisins. Par ailleurs, les tumeurs fibreuses solitaires peuvent être accompagnées d'un syndrome paranéoplasique, où la sécrétion d'une substance par la tumeur entraîne d'autres symptômes.
Les examens d'imagerie permettent de préciser la taille et la localisation exactes de la tumeur. Le diagnostic est affirmé par l'analyse de la tumeur, souvent par biopsie, qui permet également d'évaluer son potentiel d'agressivité.
Le traitement des tumeurs fibreuses solitaires est la chirurgie, qui vise à retirer la masse tumorale dans sa totalité avec une marge de sécurité.
Découverte historique
La première description d’une tumeur fibreuse solitaire remonte à 1931 ; il s’agit alors d’une tumeur au niveau de la plèvre[1] et elle est réalisée par les Américains Paul Klemperer et Coleman Rabin[2].
Les médecins anglais Arthur Purdy Stout et Margaret R. Murray ont de nouveau décrit ces tumeurs en 1942, sous le nom d'hémangiopéricytome[3],[4] et ont identifié leur nature mésenchymateuse[1]. Le nom d'hémangiopéricytome a ensuite été utilisé pour des tumeurs voisines mais considérées à tort comme distinctes des tumeurs fibreuses solitaires[5],[6] ; il désigne aujourd'hui une entité controversée de tumeurs malignes des tissus mous, proches des sarcomes[3],[7]. La classification de l'OMS des tumeurs des tissus mous a d'ailleurs supprimé l'hémangiopéricytome (dans les localisations extra-crâniennes[8]) de sa liste[9] ; les formes tissulaires ont été rattachées aux tumeurs fibreuses solitaires, et les formes périvasculaires aux tumeurs des muscles lisses[10].
De nombreux termes ont été utilisés durant le XXe siècle pour désigner les tumeurs fibreuses solitaires : tumeurs fibreuses localisées, mésothéliome fibreux localisé, mésothéliome fibreux solitaire, fibrome sous-séreux, fibrome sous-mésothélial[1]…
Épidémiologie
Les enfants comme les adultes peuvent développer des tumeurs fibreuses solitaires, mais le diagnostic est le plus souvent posé entre 50 et 70 ans[3]. L'âge est plus avancé pour les tumeurs pleurales que pour celles des autres localisations et le sex ratio est équilibré[11],[12].
Il s'agit d'une maladie rare avec seulement 900 cas publiés dans la littérature médicale en 2011[13] et 800 patients opérés aux États-Unis entre 1973 et 2012[14]. La fréquence, difficile à déterminer, est estimée à 2,8 pour 100 000[5]. Les tumeurs fibreuses solitaires sont responsables de moins de 2 % des tumeurs des tissus mous[5].
La cause est inconnue[3]. Un mécanisme de formation a cependant été décrit, avec la fusion, au sein des cellules tumorales, de deux gènes (NAB2 et STAT6[15]) entre les chromosomes 12 et 13[3],[16].
Il n'existe pas de facteur de risque environnemental ou génétique identifié[3].
Manifestations cliniques
Localisations
En raison de la rareté de la maladie, peu de grandes séries de patients sont publiées, rendant difficile l'analyse de la répartition entre les différents sites[17]. Cependant, la majorité des cas rapportés atteignent la plèvre. Cette fine membrane, qui recouvre la surface des poumons et l'intérieur de la paroi thoracique, est le siège le plus fréquent des tumeurs fibreuses solitaires[18],[19]. La fréquence estimée d'une localisation pleurale varie entre 40 %[20] et 70 %[21]. La majorité des tumeurs pleurales est développée à partir de la plèvre dite viscérale, qui tapisse le poumon (entre 65 et 80 % des tumeurs fibreuses pleurales[22]). Cependant, une étude radiologique publiée en 2015 retrouve davantage de tumeurs pariétales[23].
Les autres sites les plus touchés sont les autres séreuses de l'organisme : le péritoine et, dans une moindre mesure, le péricarde. Les tumeurs fibreuses solitaires sont cependant susceptibles de se développer dans l'ensemble des tissus mous[11], en particulier le système nerveux central[24],[20] et le rétropéritoine[19] ou le médiastin, mais aussi plus rarement le rein, le pancréas, le pelvis et l'œsophage[12],[20]. L'ensemble des localisations abdominopelviennes constitue environ un tiers des tumeurs[11]. Les tissus mous des membres, de la tête (en particulier de l'orbite, de la fosse infratemporale et des sinus) et du cou peuvent eux aussi être le siège de ces tumeurs[12].
Les localisations neurologiques concernent dans 4 cas sur 5 le cerveau, les autres atteignant la moelle épinière et intéressent surtout les adultes de plus de 30 ans (90 % des cas), mais des cas ont été décrits chez des enfants et des adolescents[12]. Les tumeurs fibreuses solitaires atteignant les méninges sont considérées comme plus proches des hémangiopéricytomes que les tumeurs des autres sites[9].
Signes cliniques
Une tumeur fibreuse solitaire progresse de manière silencieuse, sans signe clinique spécifique dans 90 % des cas[14],[17]. Lorsqu'elle est présente, la symptomatologie varie selon la localisation, et apparaît généralement lorsque la tumeur devient compressive[3],[19]. Les tumeurs fibreuses solitaires peuvent en effet être très volumineuses, en particulier dans le thorax et l'abdomen, où leur diamètre peut atteindre jusqu'à 40 cm[11]. La plupart mesurent entre 5 et 10 cm[3]. Les localisations pleurales de plus de 15 cm sont dites « géantes[25] ».
Lorsque la tumeur est superficielle et palpable, elle se présente comme une tuméfaction souple et indolore, évoluant depuis plusieurs années[3]. La réalisation plus fréquente de radiographies pulmonaires et de tomodensitométries depuis la deuxième moitié du XXe siècle permet par ailleurs de diagnostiquer plus tôt les tumeurs fibreuses solitaires pleurales, alors qu'elles sont encore asymptomatiques[13],[26].
Les tumeurs pleurales sont asymptomatiques dans 72 % des cas ; lorsque les signes cliniques sont présents, ils sont dominés par la toux, la dyspnée et les douleurs thoraciques[27]. Un épanchement pleural est parfois présent[28],[29] (environ 10 % des cas[30]). Les tumeurs abdominales se manifestent par des douleurs abdominales et une constipation, parfois une rétention d'urine, et sont parfois accompagnées d'une tuméfaction palpable[31].
En cas de localisation dans le système nerveux central, les signes cliniques dépendent de la localisation exacte et de la taille de la tumeur, et vont de la simple céphalée ou de sensations vertigineuses aux crises d'épilepsie (totale ou partielle) ou au déficit moteur et au syndrome vestibulaire, voire à une hypertension intracrânienne[12]. Les tumeurs spinales entraînent, elles, des douleurs rachidiennes, des déficits sensitifs ou moteurs, voire une myélopathie progressive et une paraplégie, selon le niveau de la compression. Les fonctions des sphincters sont généralement respectées[12].
Les tumeurs de la tête et du cou sont en règle générale peu symptomatiques et découvertes sur la palpation de la masse par le patient lui-même[32]. Les formes orbitaires peuvent en revanche entraîner un ptosis, une diplopie et une exophtalmie unilatérale[32],[33].
Si l'évolution naturelle des tumeurs fibreuses solitaires est mal connue, elle semble cependant lente, sur plusieurs années[34]. Toutefois, après plusieurs années d'évolution à bas bruit, ces tumeurs peuvent progresser rapidement, ce qui justifie un traitement rapide une fois le diagnostic posé[35].
Syndromes paranéoplasiques
On observe parfois, associé à une tumeur fibreuse solitaire, un syndrome paranéoplasique. Ce terme recouvre l'ensemble des anomalies pouvant accompagner certains cancers. Ces anomalies ne sont pas en relation directe avec la tumeur, mais sont des manifestations survenant à distance de l'endroit où se développe le cancer, par production d'une substance par la tumeur. Il s'agit de syndromes rares, surtout retrouvés dans une minorité des cancers intrathoraciques et digestifs. Le syndrome paranéoplasique accompagne parfois la tumeur dans son évolution : il la précède souvent, régresse parfois avec son traitement, disparaît avec la guérison et réapparaît en cas de rechute. Son traitement consiste à traiter le cancer responsable[36].
Le plus fréquent est le syndrome de Doege-Potter[37],[38], qui associe tumeur fibreuse solitaire et hypoglycémies. Il est présent lorsque la tumeur sécrète une hormone proche de l'insuline, appelée IGF-2, qui comme elle diminue le taux sanguin de glucose[3],[39]. Jusqu'à 80 % des tumeurs fibreuses solitaires expriment l'IGF-2[39], mais moins de 5 % des patients présentent des hypoglycémies vraies[37]. D'autres syndromes paranéoplasiques peuvent être observés, comme l'ostéoarthropathie hypertrophiante pneumique, une atteinte articulaire, dont la fréquence varie dans les séries rapportées entre 2[40] et 20 %[29]. La cause en est débattue et est rattachée à l'hypersécrétion par la tumeur soit d'acide hyaluronique, soit du facteur de croissance des hépatocytes[13].
Les syndromes paranéoplasiques sont moins fréquents dans les tumeurs atteignant le système nerveux que dans les autres localisations[12].
Bilan biologique
Il n'existe pas de marqueur sanguin dosable spécifique des tumeurs fibreuses solitaires.
Imagerie
De nombreux examens d'imagerie permettent d'explorer les tumeurs fibreuses solitaires, mais aucun ne permet de poser de diagnostic de certitude, qui est obtenue par l'analyse d'une biopsie ou de la pièce opératoire[41].
Les tumeurs fibreuses solitaires pleurales sont visualisées sur la radiographie thoracique comme des masses bien délimitées. Les clichés radiologiques standards sont cependant peu utiles pour les autres localisations[17].
En tomodensitométrie[20],[31], quelle que soit la localisation, une tumeur fibreuse solitaire apparaît comme une masse tissulaire ovoïde ou lobulée, bien délimitée, refoulant les structures voisines sans les envahir. Elle se rehausse fortement après injection de produit de contraste. Les tumeurs de petite taille sont homogènes, mais les plus volumineuses présentent des plages hétérogènes de nécrose et d'hémorragie intra-tumorale. Il n'y a pas habituellement de calcifications et, lorsqu'elles sont présentes, elles sont de petite taille[31]. Les tumeurs malignes peuvent se présenter accompagnées de nodules pulmonaires métastatiques, et tendent à être plus volumineuses et plus hétérogènes[42].
L'imagerie par résonance magnétique permet une exploration complémentaire dans certaines localisations. Elle est surtout réalisée pour les tumeurs du système nerveux central[8],[12],[43], du foie et du pelvis[31],[41]. Elle évalue mieux les différences de densité intra-tumorale et permet d'affirmer l'absence d'envahissement des tissus mous et des structures osseuses voisines[20]. Les tumeurs fibreuses solitaires ont un aspect d'isosignal en séquence T1 et d'hypersignal en T2, et se rehaussent après l'injection de gadolinium[31]. Cependant, dans le système nerveux central, elles ne présentent pas de critères caractéristiques permettant de poser un diagnostic de manière uniquement radiologique[12] et, en pré-opératoire, sont souvent confondues avec un méningiome[43].
- Tumeur fibreuse solitaire du rétropéritoine
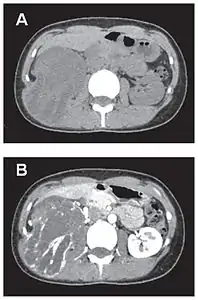 Aspect tomodensitométrique, sans (A) puis avec (B) injection de produit de contraste[44].
Aspect tomodensitométrique, sans (A) puis avec (B) injection de produit de contraste[44].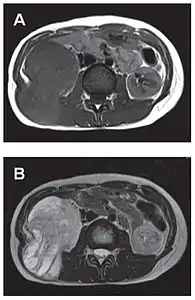 Aspect IRM, en séquence T1 (A) et T2 (B)[44].
Aspect IRM, en séquence T1 (A) et T2 (B)[44].
Le TEP scanner est utilisé afin de caractériser l'activité métabolique de la tumeur et de la différencier des tumeurs malignes. Le traceur est fixé de manière hétérogène et peu intense[41],[23].

L'échographie est rarement réalisée pour les localisations pleurales, mais peut être utile dans les autres localisations[45]. Les tumeurs fibreuses solitaires se présentent comme des masses homogènes, hypoéchogènes et bien délimitées ; le doppler permet de visualiser la vascularisation. Les tumeurs pleurales se mobilisent avec les mouvements ventilatoires[45].
L'angiographie est utilisée pour repérer la vascularisation des tumeurs intra-crâniennes. Celles-ci sont richement vascularisées par des branches artérielles naissant soit des carotides internes ou externes, soit du polygone de Willis, et il existe un blush tumoral tardif[12]. Une autre application de l'angiographie est la réalisation d'une embolisation des artères nourricières de la tumeur, afin de diminuer le saignement pendant l'intervention[13],[31],[41].
- Tumeur fibreuse solitaire du rein
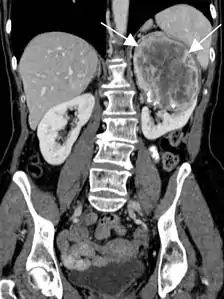 Aspect tomodensitométrique. La tumeur est repérée par les flèches[46].
Aspect tomodensitométrique. La tumeur est repérée par les flèches[46].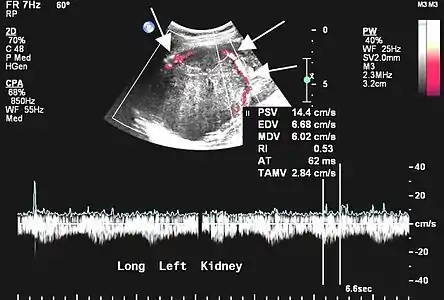 Aspect échographique. La vascularisation est visible en rouge, repérée par les flèches[46].
Aspect échographique. La vascularisation est visible en rouge, repérée par les flèches[46].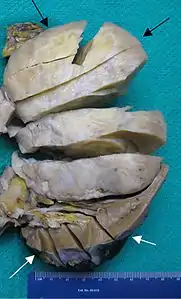 Pièce opératoire de néphrectomie : la tumeur est d'apparence rosée, repérée par les flèches noires. Le rein normal est de couleur brune, sombre, en bas de l'image et repéré par les flèches blanches[46].
Pièce opératoire de néphrectomie : la tumeur est d'apparence rosée, repérée par les flèches noires. Le rein normal est de couleur brune, sombre, en bas de l'image et repéré par les flèches blanches[46].
Démarche diagnostique

Le diagnostic clinique est difficile en raison de l'absence de signes spécifiques, et nécessite la réalisation d'examens complémentaires[47]. Si le diagnostic peut être évoqué face à une imagerie typique, le diagnostic de certitude nécessite une approche anatomopathologique de la tumeur[5]. La ponction sous scanner aide ainsi à poser le diagnostic et à orienter le traitement[13]. En effet, les diagnostics différentiels sont multiples et dépendent de la localisation de la tumeur[41]. Le plus fréquent est la fibromatose, mais toutes les tumeurs tissulaires sont concernées, en particulier l'histiocytofibrome, les sarcomes, et les tumeurs germinales[17],[48].
Au moment du diagnostic, environ 14 % des patients présentent, en plus de la tumeur principale, des localisations dans d'autres organes, considérées comme des métastases[14].
Anatomie pathologique
Aspect macroscopique
La majorité des tumeurs fibreuses solitaires pleurales (75 à 80 %) se développe à partir de la plèvre viscérale et présente un pied d'implantation plus ou moins large à la surface du poumon[27]. Ces tumeurs sont hypervascularisées[3],[13] et peuvent présenter une capsule[3]. La vascularisation, abondante et ramifiée, est dite « en corne de cerf » (staghorn en anglais)[3],[12].
Histologie
L'histologie est l'étude des tissus biologiques. Dans le cadre des tumeurs, elle s'attache à l'identification de l'architecture des tissus, mais aussi aux divers récepteurs qu'ils expriment, afin d'identifier et de classifier les tumeurs. Elle apporte le diagnostic de certitude et permet d'orienter le traitement.
Les tumeurs fibreuses solitaires sont constituées de cellules mésenchymateuses, de différenciation fibroblastique[6]. Elles sont ovoïdes ou fusiformes. Leur cytoplasme est riche en collagène[3]. L'architecture tissulaire est hétérogène, supportée par un tissu conjonctif collagénique et alterne entre des zones bien fournies en cellules et des zones plus pauvres, où l'espace intercellulaire est plus riche en acide hyaluronique[3].
L'immunohistochimie retrouve l'antigène CD34 dans 80 % des cas[30], ce qui permet de différencier les tumeurs fibreuses solitaires des mésothéliomes[3]. Les tumeurs expriment également la vimentine, mais aucune des cytokératines[30].
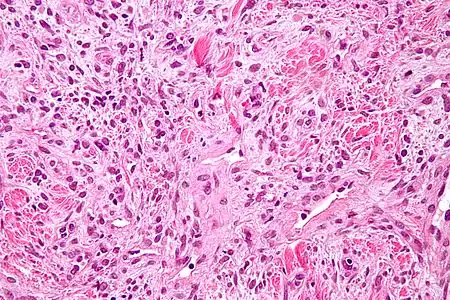 Aspect en microscopie optique (coloration H&E).
Aspect en microscopie optique (coloration H&E).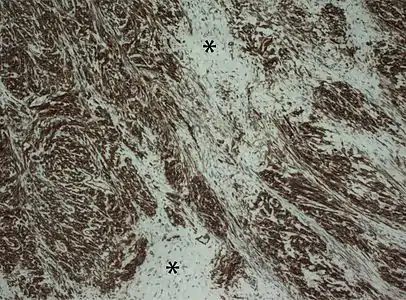 Marquage immunohistochimique des antigènes CD34, en noir. Les régions claires repérées d'un astérisque sont pauvres en cellules et riches en collagène[46].
Marquage immunohistochimique des antigènes CD34, en noir. Les régions claires repérées d'un astérisque sont pauvres en cellules et riches en collagène[46].
Il existe quelques variantes histologiques. Les tumeurs fibreuses solitaires dites « anaplasiques » ou « dédifférenciées » sont de moins bon pronostic et peuvent prendre l'aspect d'un sarcome pléomorphe. Les variantes myxoïdes et lipidiques ne modifient en revanche pas le pronostic[48]. Les différents types de fusion entre les gènes NAB2 et STAT6 pourraient être à l'origine de ces variantes histologiques[49].
Agressivité
La majorité des tumeurs fibreuses solitaires est bénigne, mais certaines présentent des critères de malignité. L'OMS classe ainsi les tumeurs fibreuses solitaires comme des tumeurs de malignité intermédiaire[10].
La proportion de tumeurs fibreuses solitaires malignes varie selon les séries de 6 %[24] à 23 %[1], voire un tiers des patients[13],[29]. Lors de l'analyse anatomo-pathologique, certaines tumeurs fibreuses solitaires présentent en effet des caractéristiques de tumeur maligne. La première définition des critères de malignité dans ces tumeurs a été réalisée par England en 1989[13]. Il identifie ainsi[22] une concentration cellulaire importante, une activité mitotique élevée, le pléomorphisme (aspect variable des cellules), et la présence d'hémorragie et de nécrose intra-tumorales. La perte d'expression de CD34 est fréquente[3]. Cependant, ces caractéristiques histologiques d'agressivité ne sont pas toujours corrélées à un plus mauvais pronostic[50], et environ 2 %[51] des formes bénignes évoluent de manière agressive[1].
Parmi les tumeurs pleurales, les formes bénignes sont plus souvent développées à partir de la plèvre viscérale, tandis que les formes malignes sont plus souvent implantées sur la plèvre pariétale[28].
Traitement
Il n'existe pas de recommandation internationale spécifique sur le traitement des tumeurs fibreuses solitaires, qui est décrit avec celui des sarcomes des tissus mous[53],[54]. Il doit être discuté en réunion de concertation pluridisciplinaire entre les chirurgiens de la région concernée par la tumeur et les spécialistes des sarcomes (oncologues et radiothérapeuthes)[5].
Chirurgie
Le traitement est chirurgical, basé sur la résection complète de la tumeur, avec de larges marges de sécurité (1 à 2 cm[51])[54]. L'opération permet généralement la guérison pour les tumeurs bénignes et la qualité des marges diminue le risque de récidive des formes malignes[21],[29].
Les tumeurs pleurales peuvent être opérées par thoracotomie ou thoracoscopie[29],[55]. Lorsqu'elles sont appendues à la plèvre viscérale, une résection atypique pulmonaire suffit ; en cas de tumeur pénétrant dans le parenchyme pulmonaire, ou très proche des bronches lobaires ou des vaisseaux, une lobectomie pulmonaire peut être nécessaire[56]. Les tumeurs développées à partir de la plèvre pariétale sont retirées avec celle-ci, et les marges de sécurité sont plus difficiles à obtenir[29].
Dans le cas des tumeurs développées à partir d'autres organes, la chirurgie est là encore considérée comme le traitement de référence[33],[57],[58].
Dans le cas des tumeurs les plus volumineuses, qui sont très vascularisées, l'intervention peut être précédée d'une embolisation des vaisseaux nourriciers de la tumeur, en radiologie interventionnelle, afin de diminuer le saignement peropératoire[13],[31].
Traitements non chirurgicaux
En cas de résection incomplète lors de la chirurgie, la radiothérapie externe peut être utilisée, mais son usage doit être discuté au cas par cas[5],[51]. Les chimiothérapies classiques ne sont pas conseillées en post-opératoire[5].
En cas de tumeur inopérable, la place des chimiothérapies est difficile à préciser[5]. Des essais ont été réalisés avec les molécules utilisées dans le traitement des sarcomes, avec une efficacité limitée. En revanche, des thérapies ciblées, en particulier l'association bévacizumab et témozolomide[59], ou le sunitinib[60], sont recommandées[53],[54]. D'autres molécules comme l'imatinib[61] ou la trabectédine[62], ont également été essayées, avec des résultats prometteurs[1]. Leur action anti-angiogénique, en diminuant la vascularisation de la tumeur, permettrait l'arrêt de sa croissance, sans pour autant en diminuer la taille[5]. La recherche se porte également sur les molécules ciblant les récepteurs de l'IGF1, exprimés par les tumeurs fibreuses solitaires[63].
Pronostic et suivi
Le pronostic des tumeurs fibreuses solitaires est excellent[13], en particulier pour les formes bénignes[29] . Selon les séries, la survie sans récidive à 5 ans est de 63 %[14] à 90 %[11],[50], et à 10 ans de 53 %[14] à 73 %[11] voire 86 %[50]. Les récidives après traitement sont le plus souvent à distance, avec le développement de métastases dans d'autres organes, principalement dans le poumon, l'os et le foie[64]. Une récidive locale est également possible, mais moins fréquente (10 % des patients[11]). Les facteurs de risque de récidive après traitement sont : un âge plus avancé au diagnostic, la taille de la tumeur, une résection incomplète de la tumeur, son stage, et les signes de malignité à l'examen anatomo-pathologique[11], [14]. La résection complète est le principal facteur sur lequel il est possible d'agir[50].
Selon la localisation, un suivi tomodensitométrique ou par résonance magnétique est préconisé, mais sans consensus sur la durée, de 10 ans pour les uns[11] et 2 ans pour les autres[51]. En 2013, Tapias et coll. ont proposé, pour les tumeurs pleurales, un score de risque de récidive basé sur un système de points[65],[66]. Un score inférieur à 3 indique un risque nul de récidive à 15 ans. Le score analyse la localisation (plèvre pariétale ou viscérale), le caractère pédiculé ou non de la tumeur, la taille (plus ou moins de 10 cm), et les signes histologiques de malignité. Le but de ce score est de guider les modalités du suivi post-opératoire[65].
Références
- (en-US) Gerald Langman, « Solitary fibrous tumor: A pathological enigma and clinical dilemma », Journal of Thoracic Disease, vol. 3, , p. 86–87 (ISSN 2077-6624, PMID 22263070, PMCID 3256512, DOI 10.3978/j.issn.2072-1439.2011.03.04, lire en ligne, consulté le ).
- (en) Paul Klemperer et Coleman B. Rabin, « Primary Neoplasms of the pleura. A report of five cases », American Journal of Industrial Medicine, vol. 22, , p. 4–31 (ISSN 1097-0274, DOI 10.1002/ajim.4700220103, lire en ligne, consulté le ).
- Thway et coll. 2016.
- (en) A. P. Stout et M. R. Murray, « Hemangiopericytoma: a vascular tumor featuring Zimmermann's pericytes », Annals of Surgery, vol. 116, , p. 26–33 (ISSN 0003-4932, PMID 17858068, PMCID 1543753, lire en ligne, consulté le ).
- (en) « Solitary fibrous tumor », sur UpToDate, (consulté le ).
- (en) « The WHO Classification of Tumours of Soft Tissue and Bone (Sarcomas) », sur Liddy Shriver Sarcoma Initiative (consulté le ).
- Perrine Marec-Bérard, « Tumeur fibreuse solitaire », sur Orphanet, (consulté le ).
- (en) Ge Wen, Meifang Li, Lijun Xu et Peiqian Hu, « Solitary fibrous tumor of the central nervous system: report of 2 cases and review of literature », International Journal of Clinical and Experimental Pathology, vol. 7, , p. 3444–3448 (ISSN 1936-2625, PMID 25031774, PMCID 4097254, lire en ligne, consulté le ).
- (en) Corinne Bouvier, Philippe Métellus, André Maues de Paula et Alexandre Vasiljevic, « Solitary Fibrous Tumors and Hemangiopericytomas of the Meninges: Overlapping Pathological Features and Common Prognostic Factors Suggest the Same Spectrum of Tumors », Brain Pathology, vol. 22, , p. 511–521 (ISSN 1750-3639, DOI 10.1111/j.1750-3639.2011.00552.x, lire en ligne, consulté le ).
- (en) C D M Fletcher, « The evolving classification of soft tissue tumours: an update based on the new WHO classification », Histopathology, vol. 48, , p. 3–12 (ISSN 1365-2559, DOI 10.1111/j.1365-2559.2005.02284.x, lire en ligne, consulté le ).
- Demicco et coll. 2012.
- Bisceglia et coll. 2011.
- Thakkar et coll. 2011.
- (en) Alimujiang Wushou, Yi-Zhou Jiang, Yi-Rong Liu et Zhi-Ming Shao, « The demographic features, clinicopathologic characteristics, treatment outcome and disease-specific prognostic factors of solitary fibrous tumor: a population-based analysis », Oncotarget, vol. 6, (ISSN 1949-2553, DOI 10.18632/oncotarget.6174, lire en ligne, consulté le ).
- (en) Juliann Chmielecki, Aimee M Crago, Mara Rosenberg et Rachael O'Connor, « Whole-exome sequencing identifies a recurrent NAB2-STAT6 fusion in solitary fibrous tumors », Nature Genetics, vol. 45, , p. 131–132 (DOI 10.1038/ng.2522, lire en ligne).
- (en) Arezoo Mohajeri, Johnbosco Tayebwa, Anna Collin et Jenny Nilsson, « Comprehensive genetic analysis identifies a pathognomonic NAB2/STAT6 fusion gene, nonrandom secondary genomic imbalances, and a characteristic gene expression profile in solitary fibrous tumor », Genes, Chromosomes and Cancer, vol. 52, , p. 873–886 (ISSN 1098-2264, DOI 10.1002/gcc.22083, lire en ligne, consulté le ).
- (en) Francis N. Musyoki, Ayoub Nahal et Thomas I. Powell, « Solitary fibrous tumor: an update on the spectrum of extrapleural manifestations », Skeletal Radiology, vol. 41, , p. 5–13 (ISSN 0364-2348 et 1432-2161, DOI 10.1007/s00256-010-1032-z, lire en ligne, consulté le ).
- S. Lamhamedi, L. Sbihi, FZ Gueddari, « La tumeur fibreuse solitaire : bénigne, vous avez dit bénigne ? » (consulté le ).
- (en) Jun Sun, Xiang-rong Yu, Bin-bin Shi et Jin Zheng, « CT features of retroperitoneal solitary fibrous tumor: report of three cases and review of the literature », World Journal of Surgical Oncology, vol. 12, , p. 324 (ISSN 1477-7819, PMID 25351104, PMCID 4282173, DOI 10.1186/1477-7819-12-324, lire en ligne, consulté le ).
- (en) Ma Zhanlong, Shi Haibin, Fang Xiangshan et Song Jiacheng, « Variable Solitary Fibrous Tumor Locations », Medicine, vol. 95, (DOI 10.1097/md.0000000000003031, lire en ligne, consulté le ).
- (en) Jason S. Gold, Cristina R. Antonescu, Cristina Hajdu et Cristina R. Ferrone, « Clinicopathologic correlates of solitary fibrous tumors », Cancer, vol. 94, , p. 1057–1068 (ISSN 0008-543X, PMID 11920476, lire en ligne, consulté le ).
- (en) D. M. England, L. Hochholzer et M. J. McCarthy, « Localized benign and malignant fibrous tumors of the pleura. A clinicopathologic review of 223 cases », The American Journal of Surgical Pathology, vol. 13, , p. 640–658 (ISSN 0147-5185, PMID 2665534, lire en ligne, consulté le ).
- (en) Yoo Kyung Yeom, Mi Young Kim, Hyun Joo Lee et Sung-Soo Kim, « Solitary Fibrous Tumors of the Pleura of the Thorax », Medicine, vol. 94, (DOI 10.1097/md.0000000000001548, lire en ligne, consulté le ).
- (en) Kyle M. Fargen, Katherine J. Opalach, Dara Wakefield et R. Patrick Jacob, « The central nervous system solitary fibrous tumor: A review of clinical, imaging and pathologic findings among all reported cases from 1996 to 2010 », Clinical Neurology and Neurosurgery, vol. 113, , p. 703–710 (DOI 10.1016/j.clineuro.2011.07.024, lire en ligne).
- (en) T. Pusiol, I. Piscioli, M. Scialpi et E. Hanspeter, « Giant benign solitary fibrous tumour of the pleura (> 15 cm): role of radiological pathological correlations in management. Report of 3 cases and review of the literature », Pathologica, vol. 105, , p. 77–82 (ISSN 0031-2983, PMID 24047032, lire en ligne, consulté le ).
- (en) Michael Briselli, Eugene J. Mark et G. Richard Dickersin, « Solitary fibrous tumors of the pleura: Eight new cases and review of 360 cases in the literature », Cancer, vol. 47, , p. 2678–2689 (ISSN 1097-0142, DOI 10.1002/1097-0142(19810601)47:113.0.CO;2-9, lire en ligne, consulté le ).
- (en) Serkan Enon, Cabir Yuksel, Ayten Kayi Cangir et Sibel Percinel, « Benign Localized Fibrous Tumor of the Pleura: Report of 25 New Cases », The Thoracic and Cardiovascular Surgeon, vol. 60, , p. 468–473 (ISSN 0171-6425, DOI 10.1055/s-0031-1295519, lire en ligne, consulté le ).
- (en) George Rakovich, Maxime Laflamme, Denise Ouellette et Gilles Beauchamp, « Solitary fibrous tumour of the pleura: A case report », Canadian Respiratory Journal, vol. 17, , p. 113–114 (ISSN 1916-7245, PMID 20617210, PMCID 2900141, lire en ligne, consulté le ).
- Magdeleinat et coll. 2002.
- (en) M. Suter, S. Gebhard, M. Boumghar et N. Peloponisios, « Localized fibrous tumours of the pleura: 15 new cases and review of the literature », European Journal of Cardio-Thoracic Surgery, vol. 14, , p. 453–459 (ISSN 1010-7940 et 1873-734X, DOI 10.1016/S1010-7940(98)00213-9, lire en ligne, consulté le ).
- Li et coll. 2014.
- (en) Yu Liu, KaiCheng Li, Huimin Shi et XiaoFeng Tao, « Solitary fibrous tumours in the extracranial head and neck region: correlation of CT and MR features with pathologic findings », La radiologia medica, vol. 119, , p. 910–919 (ISSN 0033-8362 et 1826-6983, DOI 10.1007/s11547-014-0409-9, lire en ligne, consulté le ).
- (en) Crystal P. Le, Scott Jones et Alejandra A. Valenzuela, « Orbital Solitary Fibrous Tumor: A Case Series with Review of the Literature », Orbit, vol. 33, , p. 145–151 (ISSN 0167-6830, DOI 10.3109/01676830.2013.853806, lire en ligne, consulté le ).
- (en) Hyun Woo Jeon, Soon Seog Kwon et Young-Du Kim, « Malignant solitary fibrous tumor of the pleura slowly growing over 17 years: case report », Journal of Cardiothoracic Surgery, vol. 9, , p. 113 (ISSN 1749-8090, DOI 10.1186/1749-8090-9-113, lire en ligne, consulté le ).
- (en) Tamio Okimoto, Yasushi Horimasu, Shunichi Hamaguchi et Akihisa Sutani, « Solitary fibrous tumor with rapid progression after 16 years' follow up », Internal Medicine (Tokyo, Japan), vol. 53, , p. 617–621 (ISSN 1349-7235, PMID 24633034, lire en ligne, consulté le ).
- Larousse Médical, « Syndrome paranéoplasique », sur www.larousse.fr (consulté le )
- (en) Haider Zafar, Chris H. Takimoto et Geoffrey Weiss, « Doege-Potter syndrome: hypoglycemia associated with malignant solitary fibrous tumor », Medical Oncology (Northwood, London, England), vol. 20, , p. 403–408 (ISSN 1357-0560, PMID 14716039, DOI 10.1385/MO:20:4:403, lire en ligne, consulté le ).
- (en) Wen Meng, Hong-Hong Zhu, Hu Li et Guoqing Wang, « Solitary fibrous tumors of the pleura with Doege-Potter syndrome: a case report and three-decade review of the literature », BMC research notes, vol. 7, , p. 515 (ISSN 1756-0500, PMID 25113505, PMCID 4267432, DOI 10.1186/1756-0500-7-515, lire en ligne, consulté le ).
- (en) Sonja E. Steigen, David F. Schaeffer, Robert B. West et Torsten O. Nielsen, « Expression of insulin-like growth factor 2 in mesenchymal neoplasms », Modern Pathology, vol. 22, , p. 914–921 (ISSN 0893-3952, DOI 10.1038/modpathol.2009.48, lire en ligne, consulté le ).
- (en) Sook Hwan Sung, Jee-Won Chang, Jhingook Kim et Kyung Soo Lee, « Solitary Fibrous Tumors of the Pleura: Surgical Outcome and Clinical Course », The Annals of Thoracic Surgery, Elsevier, vol. 79, no 1, , p. 303-307 (lire en ligne).
- (en) Alampady K. Shanbhogue, Srinivasa R. Prasad, Naoki Takahashi et Raghunandan Vikram, « Somatic and Visceral Solitary Fibrous Tumors in the Abdomen and Pelvis: Cross-sectional Imaging Spectrum », RadioGraphics, vol. 31, , p. 393–408 (ISSN 0271-5333, DOI 10.1148/rg.312105080, lire en ligne, consulté le ).
- (en) Sun Wha Song, Jung Im Jung, Kyo Young Lee et Mi-Young Kim, « Malignant solitary fibrous tumor of the pleura: computed tomography-pathological correlation and comparison with computed tomography of benign solitary fibrous tumor of the pleura », Japanese Journal of Radiology, vol. 28, , p. 602–608 (ISSN 1867-1071 et 1862-5274, DOI 10.1007/s11604-010-0484-3, lire en ligne, consulté le ).
- (en) Frédéric Clarençon, Fabrice Bonneville, Audrey Rousseau et Damien Galanaud, « Intracranial solitary fibrous tumor: Imaging findings », European Journal of Radiology, vol. 80, , p. 387–394 (DOI 10.1016/j.ejrad.2010.02.016, lire en ligne).
- (en) Takeo Nomura, Ryuta Satoh, Kenji Kashima et Mutsushi Yamasaki, « A case of large solitary fibrous tumor in the retroperitoneum », Clinical Medicine. Case Reports, vol. 2, , p. 21–25 (ISSN 1178-6450, PMID 24179368, PMCID 3785379, lire en ligne, consulté le ).
- (en) Mitsuaki Sekiya, Kaku Yoshimi, Keiko Muraki et Kenji Suzuki, « Solitary fibrous tumor of the pleura: Ultrasonographic imaging findings of 3 cases », Respiratory Investigation, vol. 51, , p. 200–204 (DOI 10.1016/j.resinv.2013.04.001, lire en ligne).
- (en) Dzmitry Fursevich, Edward Derrick, Matthew C O'Dell, Swetha Vuyyuru, Jeremy Burt, « Solitary Fibrous Tumor of the Kidney: A Case Report and Literature Review », Cureus, (lire en ligne).
- (en) Ademola Adegoke Aremu, Olusola Adetunji Oyedeji, Christianah Mopelola Asaleye et Victor Adebayo Adetiloye, « An elusive chest coin in an african child: a pleural fibroma's long, tortuous path to freedom », Pan African Medical Journal, vol. 2, (DOI 10.11604/pamj.2013.14.16.1874, lire en ligne, consulté le ).
- « Tumeur fibreuse solitaire des tissus mous », sur Anabible, (consulté le ).
- (en) Hui-Chun Tai, I.-Chieh Chuang, Tse-Ching Chen et Chien-Feng Li, « NAB2–STAT6 fusion types account for clinicopathological variations in solitary fibrous tumors », Modern Pathology, vol. 28, , p. 1324–1335 (ISSN 0893-3952, DOI 10.1038/modpathol.2015.90, lire en ligne, consulté le ).
- (en) Guillaume Boddaert, Patrice Guiraudet, Bertrand Grand et Nicolas Venissac, « Solitary fibrous tumors of the pleura: a poorly defined malignancy profile », The Annals of Thoracic Surgery, vol. 99, , p. 1025–1031 (ISSN 1552-6259, PMID 25620590, DOI 10.1016/j.athoracsur.2014.10.035, lire en ligne, consulté le ).
- (en) Marc de Perrot, Stefan Fischer, Marie-Anne Bründler et Yasuo Sekine, « Solitary fibrous tumors of the pleura », The Annals of Thoracic Surgery, vol. 74, , p. 285–293 (ISSN 0003-4975, PMID 12118790, lire en ligne, consulté le ).
- (en) Laura Schirosi, Sylvie Lantuejoul, Alberto Cavazza et Bruno Murer, « Pleuro-pulmonary Solitary Fibrous Tumors », The American Journal of Surgical Pathology, vol. 32, , p. 1627–1642 (DOI 10.1097/pas.0b013e31817a8a89, lire en ligne, consulté le ).
- (en) « NCCN guidelines : soft tissue sarcoma » (consulté le ).
- (en) Xavier Garcia del Muro, Enrique de Alava, Vicenç Artigas et Silvia Bague, « Clinical practice guidelines for the diagnosis and treatment of patients with soft tissue sarcoma by the Spanish group for research in sarcomas (GEIS) », Cancer Chemotherapy and Pharmacology, vol. 77, , p. 133–146 (ISSN 1432-0843, PMID 26563256, DOI 10.1007/s00280-015-2809-5, lire en ligne, consulté le ).
- (en) Jun Liu, Chengjie Cai, Daoyuan Wang et Hanzhang Chen, « Video-Assisted Thoracoscopic Surgery (VATS) for Patients with Solitary Fibrous Tumors of the Pleura », Journal of Thoracic Oncology, vol. 5, , p. 240–243 (ISSN 1556-0864, DOI 10.1097/jto.0b013e3181c6b6b2, lire en ligne, consulté le ).
- (en) Karen M. Harrison–Phipps, Francis C. Nichols, Cathy D. Schleck et Claude Deschamps, « Solitary fibrous tumors of the pleura: Results of surgical treatment and long-term prognosis », The Journal of Thoracic and Cardiovascular Surgery, vol. 138, , p. 19–25 (DOI 10.1016/j.jtcvs.2009.01.026, lire en ligne).
- (en) Nazih Khater, Raja Khauli, Mohammad Shahait et Jad Degheili, « Solitary Fibrous Tumors of the Kidneys: Presentation, Evaluation, and Treatment », Urologia Internationalis, vol. 91, , p. 373–383 (DOI 10.1159/000354394, lire en ligne, consulté le ).
- (en) Paula Novais, Carlos Robles-Medranda, Vera Lucia Pannain et Daniel Barbosa, « Solitary fibrous liver tumor: is surgical approach the best option? », Journal of gastrointestinal and liver diseases: JGLD, vol. 19, , p. 81–84 (ISSN 1841-8724, PMID 20361081, lire en ligne, consulté le ).
- (en) Min S. Park, Shreyaskumar R. Patel, Joseph A. Ludwig et Jonathan C. Trent, « Activity of temozolomide and bevacizumab in the treatment of locally advanced, recurrent, and metastatic hemangiopericytoma and malignant solitary fibrous tumor », Cancer, vol. 117, , p. 4939–4947 (ISSN 1097-0142, DOI 10.1002/cncr.26098, lire en ligne, consulté le ).
- (en) S. Stacchiotti, T. Negri, M. Libertini et E. Palassini, « Sunitinib malate in solitary fibrous tumor (SFT) », Annals of oncology: official journal of the European Society for Medical Oncology / ESMO, vol. 23, , p. 3171–3179 (ISSN 1569-8041, PMID 22711763, DOI 10.1093/annonc/mds143, lire en ligne, consulté le ).
- (en) Tommaso De Pas, Francesca Toffalorio, Piergiuseppe Colombo et Giuseppe Trifirò, « Brief report: activity of imatinib in a patient with platelet-derived-growth-factor receptor positive malignant solitary fibrous tumor of the pleura », Journal of Thoracic Oncology: Official Publication of the International Association for the Study of Lung Cancer, vol. 3, , p. 938–941 (ISSN 1556-1380, PMID 18670317, DOI 10.1097/JTO.0b013e3181803f08, lire en ligne, consulté le ).
- (en) J. Khalifa, M. Ouali, L. Chaltiel et S. Le Guellec, « Efficacy of trabectedin in malignant solitary fibrous tumors: a retrospective analysis from the French Sarcoma Group », BMC cancer, vol. 15, , p. 700 (ISSN 1471-2407, PMID 26472661, PMCID 4608145, DOI 10.1186/s12885-015-1697-8, lire en ligne, consulté le ).
- (en) Richard Quek, Qian Wang, Jeffrey A. Morgan et Geoffrey I. Shapiro, « Combination mTOR and IGF-1R inhibition: phase I trial of everolimus and figitumumab in patients with advanced sarcomas and other solid tumors », Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, vol. 17, , p. 871–879 (ISSN 1078-0432, PMID 21177764, DOI 10.1158/1078-0432.CCR-10-2621, lire en ligne, consulté le ).
- (en) Zhengrong Wu, Hongjun Yang, Desheng Weng et Yanqing Ding, « Rapid recurrence and bilateral lungs, multiple bone metastasis of malignant solitary fibrous tumor of the right occipital lobe: report of a case and review », Diagnostic Pathology, vol. 10, , p. 91 (ISSN 1746-1596, PMID 26155787, PMCID 4495700, DOI 10.1186/s13000-015-0318-9, lire en ligne, consulté le ).
- (en) Luis F. Tapias, Mari Mino-Kenudson, Hang Lee et Cameron Wright, « Risk factor analysis for the recurrence of resected solitary fibrous tumours of the pleura: a 33-year experience and proposal for a scoring system », European Journal of Cardio-Thoracic Surgery, vol. 44, , p. 111–117 (ISSN 1010-7940 et 1873-734X, DOI 10.1093/ejcts/ezs629, lire en ligne, consulté le ).
- (en) Luis F. Tapias, Olaf Mercier, Maria R. Ghigna et Benoit Lahon, « Validation of a Scoring System to Predict Recurrence of Resected Solitary Fibrous Tumors of the Pleura », Chest, vol. 147, , p. 216–223 (DOI 10.1378/chest.14-1180, lire en ligne, consulté le ).
Annexes
Bibliographie
Articles généraux :
- (en) Khin Thway, Wen Ng, Jonathan Noujaim et Robin L. Jones, « The Current Status of Solitary Fibrous Tumor: Diagnostic Features, Variants, and Genetics », International Journal of Surgical Pathology, vol. 24, , p. 281–292 (ISSN 1940-2465, PMID 26811389, DOI 10.1177/1066896915627485, lire en ligne, consulté le ) ;
- (en) Elizabeth G. Demicco, Min S. Park, Dejka M. Araujo et Patricia S. Fox, « Solitary fibrous tumor: a clinicopathological study of 110 cases and proposed risk assessment model », Modern Pathology, vol. 25, , p. 1298–1306 (ISSN 0893-3952, DOI 10.1038/modpathol.2012.83, lire en ligne, consulté le ).
Par localisation :
- plèvre :
- (en) Rohan G. Thakkar, Sumeet Shah, Amol Dumbre et Mukta A. Ramadwar, « Giant solitary fibrous tumour of pleura -an uncommon intrathoracic entity- a case report and review of the literature », Annals of Thoracic and Cardiovascular Surgery: Official Journal of the Association of Thoracic and Cardiovascular Surgeons of Asia, vol. 17, , p. 400–403 (ISSN 2186-1005, PMID 21881330, lire en ligne, consulté le ) ;
- (en) Pierre Magdeleinat, Marco Alifano, Antonio Petino et Jean Philippe Le Rochais, « Solitary fibrous tumors of the pleura: clinical characteristics, surgical treatment and outcome », European Journal of Cardio-Thoracic Surgery, vol. 21, , p. 1087–1093 (ISSN 1010-7940, PMID 12048090, lire en ligne, consulté le ) ;
- abdoment et pelvis :
- (en) Xue-Ming Li, Jing Reng, Peng Zhou, Ying Cao, Zhu-Zhong Cheng, Yan Xiao et Guo-Hui Xu, « Solitary fibrous tumors in abdomen and pelvis: Imaging characteristics and radiologic-pathologic correlation », World Journal of Gastroenterology, vol. 20, (PMID 24803820, PMCID 4009542, DOI 10.3748/wjg.v20.i17.5066, lire en ligne, consulté le ) ;
- système nerveux central :
- (en) Michele Bisceglia, Carlos Galliani, Giuseppe Giannatempo et Walter Lauriola, « Solitary Fibrous Tumor of the Central Nervous System », Advances In Anatomic Pathology, vol. 18, , p. 356–392 (DOI 10.1097/pap.0b013e318229c004, lire en ligne, consulté le ).
Articles connexes
- Autres tumeurs des tissus mous :
- Localisations fréquentes :
Liens externes
- H. Bégueret, « Tumeur fibreuse solitaire », sur Respir.com, (consulté le ).
- « Tumeur fibreuse solitaire des tissus mous », sur Anabible, (consulté le ).
 Portail de la médecine
Portail de la médecine