Western blot
Le western blot (également appelé transfert de protéines ou buvardage de western ou encore technique des immuno-empreintes)[1], est une méthode de biologie moléculaire permettant la détection et l'identification de protéines spécifiques dans un échantillon biologique (sérum ou autre extrait ou homogénat tissulaire) à l'aide d'anticorps dirigés contre ces protéines que l'on souhaite détecter. Le western blot permet ainsi de visualiser des protéines particulières dans un mélange complexe. C'est l'une des techniques analytiques de transfert sur membrane utilisées en biochimie et en biologie moléculaire.
Elle est parfois utilisée comme un outil de diagnostic complémentaire pour mettre en évidence une protéine particulière (protéine virale, par exemple) dans le sérum d'un patient.
Technique
Cette technique, née des progrès de la protéomique, de la biologie moléculaire et de l'Immunofluorescence, utilise l'électrophorèse sur gel de polyacrylamide pour séparer des protéines, préalablement dénaturées, selon leur taille.
Ces protéines sont ensuite transférées depuis le gel sur une membrane (typiquement en nitrocellulose), où elles sont exposées à un anticorps spécifique de la protéine d'intérêt. Il est possible grâce à cette technique de détecter la présence d'une protéine dans un tissu, d'évaluer sa taille, sa concentration, les variations de cette concentration, effectuer des comparaisons de concentrations entre différents groupes, etc. D'autres techniques utilisant les anticorps permettent la détection de la protéine dans les cellules après fixation (immunocytochimie) et dans les tissus (immunohistochimie).
La méthode fut mise au point dans le laboratoire de George Stark à Stanford. Le nom du western blot, donné à la technique par W. Neal Burnette[2], est un jeu de mot à partir de la technique du Southern blot ou transfert de Southern, technique de détection d'ADN nommée d'après son inventeur, Edwin Southern et non d'après le point cardinal. La détection d'ARN est appelée northern blot ou transfert de northern. Toutes ces techniques dérivent leur nom de l'étape de transfert sur membrane, comparée à une empreinte sur buvard (blot = « tache » en anglais).
Différentes étapes du transfert de western

Préparation des échantillons
Les échantillons (d'un tissu ou d'une culture cellulaire) sont rapidement refroidis, voire réfrigérés (en dessous de 0 °C). Ils sont homogénéisés par sonication (utilisation d'ultra-sons pour rompre les membranes), contrainte mécanique ou simplement lysés par utilisation de tampons à haute concentration en sels. Il en résulte un homogénat de tous les compartiments cellulaires, pouvant être utilisé tel quel ou être soumis à plusieurs étapes de centrifugation différentielle afin d'isoler les fractions cytosolique, nucléaire et membranaire. L'échantillon est ensuite traité de façon à recueillir un taux constant de protéines à partir de chaque échantillon différent. Cela implique un dosage des protéines par la méthode du Biuret ou du bleu de Coomassie (Méthode de Bradford).
Les échantillons sont ensuite bouillis de 1 à 5 minutes dans une solution tampon d'électrophorèse (par exemple le tampon de Laemmli), contenant une substance tampon, généralement du Tris, un colorant, un composant sulfhydryl (typiquement du beta-mercaptoéthanol ou du dithiothréitol ou plus simplement DTT), un détergent anionique lipophile (sodium dodécyl sulfate ou SDS) et du glycérol pour diminuer la poussée d'Archimède. La poussée d'Archimède entraine, un corps plongé dans un fluide, verticalement vers le haut.
L'ébullition dénature les protéines en brisant les faibles liaisons intramoléculaires, ce qui a pour conséquence de les dérouler complètement. Le SDS leur procure alors un environnement riche en charges négatives afin de les solvater et prévenir la précipitation, et le composant sulfhydryl empêche la regénération des ponts disulfure. Le glycérol augmente la densité de l'échantillon par rapport au tampon dans la partie supérieure du réservoir du gel, facilitant la mise en place des échantillons qui descendront plus facilement au fond des compartiments du gel.
Électrophorèse sur gel
Les protéines de l'échantillon sont séparées selon leur taille par électrophorèse sur gel, dont la composition varie en fonction du laboratoire, du poids moléculaire des protéines d'intérêt et des tampons disponibles. Les gels de polyacrylamide sont les plus fréquents. Les protéines ne traversant le gel que dans une dimension (du haut vers le bas), les échantillons sont chargés l'un à côté de l'autre dans des puits formés dans le gel. Les protéines sont séparées par masse en « bandes » dans chaque « couloir » formé sous les puits. Un couloir est réservé à un « marqueur » ou « échelle standard », une mixture de protéines possédant des poids moléculaires définis disponibles dans le commerce.
Il est également possible d'employer un gel 2D qui, à partir d'un seul échantillon, permet de faire migrer les protéines dans deux dimensions. Les protéines sont alors séparées par leur point isoélectrique (c'est-à-dire le pH auquel leur charge nette est neutre) dans la première dimension, et selon leur poids dans la seconde.
La composition d'un tampon d'électrophorèse (running buffer) pour western blot est d'un volume de tampon TGS (Tris-glycine-SDS) dilué 10 fois dans 9 volumes d'eau distillée.
Transfert sur membrane

Afin de rendre les protéines accessibles à la détection par anticorps, elles sont transférées depuis le gel sur une membrane de nitrocellulose ou de PVDF. La membrane est placée face-à-face avec le gel, et un courant électrique est appliqué aux grandes plaques sur l'un des deux côtés. Les protéines chargées migrent depuis le gel vers la membrane en conservant l'organisation relative qu'elles avaient dans le gel. Il résulte de ce transfert que les protéines sont exposées sur une surface mince, ce qui facilite les étapes de détection ultérieures. Tant les membranes de nitrocellulose que de PVDF sont « collantes », liant des protéines de manière non spécifique (c'est-à-dire qu'elles lient toutes les protéines présentes dans l'échantillon de la même façon). La fixation des protéines à la membrane se fait grâce à des interactions hydrophobes et ioniques entre la membrane et les protéines. Bien que les membranes de nitrocellulose soient moins chères que celles en PVDF, elles sont moins solides et ne peuvent être employées plusieurs fois. À la différence de celles en nitrocellulose, les membranes de PVDF doivent être imbibées de méthanol ou d'isopropanol à 100 % avant leur utilisation
La composition pour un litre d'un tampon de transfert type est de :
Coloration des protéines
Cette étape visualise la protéine totale qui a été transférée avec succès à la membrane. La coloration des protéines permet à l'utilisateur de vérifier l'uniformité du transfert de protéines et d'effectuer une normalisation ultérieure de la protéine cible avec la quantité réelle de protéines par voie. La normalisation avec le soi-disant "contrôle interne" a été basée sur l'immunocoloration des protéines constitutifs ou des protéines de structure (comme l'actine ou la tubuline) dans la procédure classique, mais se dirige vers la coloration des protéines totales récemment, en raison des avantages multiples.[4] Au moins sept approches différentes pour la coloration des protéines totales ont été décrites pour la normalisation des Western blot: Ponceau S, stain-free technique, Sypro Ruby, Epicocconone, Coomassie R-350, Amido Black et Cy5[4]. Afin d'éviter le bruit du signal, la coloration des protéines doit être effectuée avant le blocage de la membrane. Néanmoins, des colorations post-anticorps ont également été décrites.[5]
Blocage
La membrane ayant été choisie pour ses propriétés de liaison non spécifique, comme tant la protéine-cible que les anticorps sont des protéines, des précautions doivent être prises pour minimiser les interactions entre membrane et anticorps. Le blocage des sites d'interactions non spécifiques entre la membrane et les anticorps est réalisé en plongeant la membrane dans une solution diluée de protéines - le plus souvent de l'albumine de sérum bovin ou ASB (BSA en anglais) ou du lait sans matières grasses (lait dilué à 5 % - 5 g pour 100 ml) - en présence de détergent, typiquement du Tween 20), pendant une heure à température ambiante.
Les protéines dans la solution diluée se lient à la membrane dans tous les sites non occupés par la protéine-cible.
De la sorte, lorsque les anticorps sont appliqués lors de l'étape suivante, ils ne peuvent (idéalement) plus s'attacher à la membrane que sur les sites de liaison de la protéine-cible, ce qui réduit le « bruit de fond » dans le produit final du transfert de western, donne des résultats plus clairs et élimine les faux positifs.
Détection
Au cours de la détection, la membrane est « sondée » pour la protéine d'intérêt avec des anticorps, liés ensuite à une enzyme émettant un signal photométrique ou colorimétrique, ou bien des photons. Pour plusieurs raisons, ceci se produit classiquement en deux étapes, bien que des méthodes en une étape soient disponibles pour certaines applications.
Anticorps primaire
Les anticorps sont générés par inoculation à l'animal (généralement un lapin ou une chèvre) ou exposition d'une culture de cellules immunitaires à la protéine d'intérêt dans son intégralité ou seulement sur l'une de ses fractions (épitope). Toutefois, les protéines ayant été dénaturées lors de l'électrophorèse sur gel, il faut utiliser des anticorps qui reconnaissent spécifiquement la protéine dénaturée, et non la protéine native.
La réponse immunitaire normale est dans ce cas exploitée afin de générer des anticorps qui sont récoltés et utilisés comme outils de détection possédant à la fois une bonne sensibilité et une bonne spécificité, se liant directement à la protéine - d'où leur appellation d'anticorps « primaire ». Quelques anticorps monoclonaux, beaucoup plus difficiles à réaliser et dont l'affinité est beaucoup plus élevée peuvent également être utilisés en transfert de western.
Après le blocage, une solution diluée d'anticorps primaire (généralement comprise entre 0,5 et 5 micros grammes/ml) est incubée avec la membrane sous agitation modérée. La solution se compose typiquement d'un tampon salin proche du pH neutre (généralement, une faible quantité de chlorure de sodium), d'un petit pourcentage de détergent, et parfois de protéines (ASB ou lait 5 %) diluées. La solution d'anticorps et la membrane peuvent être scellées dans un sachet en plastique et incubées ensemble pour une durée allant de 30 minutes à une nuit. Elle peut aussi être incubée à différentes températures, les températures plus élevées étant associées à plus de liaisons plus spécifiques.
Anticorps secondaire
Après rinçage de la membrane afin d'enlever les anticorps primaires non liés, celle-ci est exposée à un autre anticorps, dirigé contre une portion espèce-spécifique de l'anticorps primaire. Cet anticorps est connu comme anticorps « secondaire », et tend à être référé, du fait de ses propriétés de ciblage, comme « anti-souris », « anti-chèvre », « anti-lapin », etc. Les anticorps sont de source animale (mais généralement, d'espèces différentes de l'espèce de l'anticorps primaire), ou de cultures d'hybridomes d'origine animale ; un anticorps anti-souris se liera à pratiquement tout anticorps primaire d'origine murine, ce qui permet de réaliser des économies en laissant le laboratoire partager une seule source d'anticorps secondaire, et permet des résultats plus reproductibles. L'anticorps secondaire est généralement lié à la biotine ou à une enzyme qui permet l'identification visuelle de la protéine étudiée sur la membrane, comme une phosphatase alcaline ou la peroxydase de raifort. Cette étape confère un avantage, en ce que plusieurs anticorps secondaires se lieront à un anticorps primaire, permettant d'améliorer le signal.
Le plus communément, un anticorps secondaire lié à la peroxydase de raifort (horseradish peroxidase) est utilisé en conjonction avec un agent luminescent, et le produit de la réaction émet une luminescence proportionnelle à la concentration en protéine. Un film photographique sensible est placé contre la membrane et, sous l'exposition de la lumière due à la réaction, crée une image des anticorps liés à la membrane. Plus souvent maintenant, une caméra à CCD est utilisée en place des films photographiques.
Comme pour les procédures d'ELISPOT et ELISA, l'enzyme peut être fournie avec une molécule-substrat qui sera converti par l'enzyme pour émettre un produit de réaction coloré, visible sur la membrane.
Une troisième possibilité consiste à utiliser un marqueur radioactif plutôt qu'une enzyme couplée à l'anticorps secondaire, par exemple en marquant une protéine liant les anticorps, telle que la protéine A du staphylocoque avec un isotope radioactif de l'iode. Cependant, les autres méthodes étant plus sûres, plus rapides et moins chères, cette méthode est plus ou moins tombée en désuétude, mais demeure parfois utilisée dans certaines circonstances.
Méthode en une étape
Lorsque la technique est apparue, le processus de détection était réalisé en deux étapes, du fait de la relative facilité de production des anticorps primaires et secondaires en deux procédés distincts. Cela permet aux chercheurs et fournisseurs une certaine souplesse à l'emploi, et ajoute une étape d'amplification du signal au processus de détection. Toutefois, depuis l'arrivée d'analyses de protéines à haut rendement et à marge de détection plus basse, c'est-à-dire détectant des protéines à très faible concentration, il est devenu intéressant de développer des systèmes de sondage en une étape unique qui permettront un gain de temps et une économie de matière première. Ces systèmes nécessitent un anticorps de détection pouvant à la fois détecter la protéine d'intérêt et émettre un signal détectable, lesquelles sont souvent disponibles pour des marqueurs de protéine connus. La sonde primaire est incubée avec la membrane à la manière de l'anticorps primaire dans le procédé en deux temps, et est ensuite prête pour la détection directe après une série d'étapes de rinçages.
Analyse
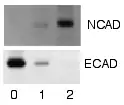
Après rinçage des anticorps secondaires non liés, le western blot est prêt pour la détection des sondes marquées et liées à la protéine d'intérêt. En pratique, tous les transferts de western ne révèlent pas la protéine sur une bande donnée de la membrane, les gels n'étant pas complètement exempts de protéines après avoir été épongés. Une approximation sur la taille est réalisée en comparant les bandes marquées à celles des marqueurs de la gamme étalon chargé durant l'électrophorèse dans un puits à part. Le processus est répété pour une protéine de structure, comme l'actine ou la tubuline, dont la concentration ne devrait pas varier entre les échantillons (contrôle interne). La concentration de la protéine-cible est indexée à celle de la protéine servant de contrôle interne, afin de normaliser les expériences. Cette pratique permet la correction par rapport au taux de protéines total sur la membrane, en cas d'erreur ou de transfert incomplet.
Détection colorimétrique
Cette méthode est tributaire, lors de l'incubation du transfert de western, de la présence d'un substrat qui réagit au déclencheur présent sur l'anticorps secondaire (comme la phosphatase alcaline). Cela convertit un colorant soluble en une forme insoluble, de couleur différente, qui précipite à côté de l'enzyme, teintant donc la membrane de nitrocellulose. Le développement du transfert de western est alors arrêté par rinçage du colorant soluble. La concentration de protéine est évaluée par densitométrie, évaluation de l'intensité de la bande ou par spectrophotométrie.
Détection par chimioluminescence
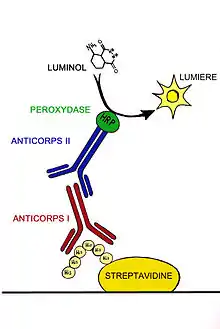
Cette méthode nécessite, lors de l'incubation du transfert de western, la présence d'un substrat qui émet de la lumière après exposition au déclencheur présent sur l'anticorps secondaire. La lumière émise sert à impressionner un film photographique, ou plus récemment, par des caméras CCD qui restituent une image numérique du transfert de western. L'image est analysée par densitométrie, qui évalue le taux relatif de marquage de la protéine, et quantifie les résultats en ce qui concerne la densité optique. Des logiciels permettent une analyse plus poussée des données, comme l'analyse du poids moléculaire si les standards appropriés sont utilisés. Cette méthode appelée détection améliorée de la chimiluminescence (enhanced chemiluminescent, ECL) est considérée comme l'une des méthodes de détection les plus sensibles pour l'analyse des western blots.
Détection par autoradiographie
Le marquage radioactif ne nécessite pas de substrat enzymatique, mais permet l'utilisation de films utilisés en médecine pour l'imagerie à rayons X. Le film est directement placé sur la membrane, qui se développe lors de son exposition au marqueur et crée des régions sombres, lesquelles correspondent aux bandes de la protéine d'intérêt (cf. illustration). L'importance des méthodes de détection par radioactivité est en déclin, du fait de son coût et de techniques plus sûres comme l'ECL.
Détection par fluorescence
La sonde couplée à l'anticorps est excitée par un rayon monochromatique et l'émission qui en résulte est alors détectée par un photosenseur, par exemple une caméra CCD équipée des filtres en transmission appropriés, qui restitue une image numérique du transfert de western et permet une analyse plus fine telle que l'analyse du poids moléculaire ou quantitative du transfert de western. La fluorescence est considérée comme d'un niveau à peu près équivalent à la chimiluminescence pour l'analyse des transferts de western. Elle nécessite un outillage plus coûteux toutefois.
Différence entre fluorescence et chimiluminescence
Bien que de mécanisme photophysique similaire, chimiluminescence et fluorescence ne sont pas la même chose :
- la fluorescence fait référence à l'excitation d'une molécule depuis un état stable vers un état excité, et son retour à l'état basal par émission d'une radiation dans le spectre électromagnétique d'une longueur d'onde donnée, et spécifique de la molécule.
- la chimiluminescence fait référence à la situation dans laquelle une molécule est formée dans un état excité, en tant que produit d'une réaction chimique, et retombe vers un état basal avec émission d'une radiation dans le spectre électromagnétique d'une longueur d'onde donnée.
Sondage secondaire
L'une des plus grandes différences entre les membranes en nitrocellulose et celles en PVDF est liée à leur capacité de supporter l'« arrachage » (stripping) d'anticorps et la réutilisation des membranes pour des sondages par d'autres anticorps. Bien qu'il existe des protocoles bien établis pour la réutilisation des membranes de nitrocellulose, le PVDF, plus épais, permet de réaliser ces manœuvres en toute sécurité et facilité, et davantage d'utilisations ultérieures avant d'être, tel un palimpseste, recouvert de signaux parasites. Une autre différence importance est que le PVDF, contrairement à la nitrocellulose, doit être trempé dans de l'éthanol à 95 % ou de l'isopropanol avant usage. Les membranes en PVDF tendent aussi à être plus épaisses et plus résistantes aux dommages physiques liés à leur utilisation normale.
Applications médicales en diagnostic
- Les tests VIH de confirmation emploient la méthode du transfert de western afin de détecter un anticorps anti-VIH dans un échantillon de sérum. Des protéines de cellules que l'on sait infectées par le VIH sont séparées et transférées sur membrane comme décrit ci-avant. Le sérum à tester est appliqué. L'étape d'incubation dans l'anticorps primaire ; les anticorps libres sont éliminés par rinçage de la membrane, et un anticorps dirigé contre les protéines humaines secondaires associé à une enzyme ou un chromophore est ajouté. Les bandes marquées indiquent ensuite les protéines contre lesquelles le sérum du patient contient des anticorps.
- Le transfert de western est également utilisé pour le test de confirmation de l'ESB (dite « maladie de la vache folle »).
- Certaines formes de détection de la maladie de Lyme utilisent le transfert de western.
Notes et références
- Voir l'article Transfert (biologie moléculaire) pour un complément sur la terminologie française.
- (en) W. Neal Burnette., « Western blotting": electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. », Analytical Biochemistry, vol. 112, no 2, , p. 195-203 (PMID 6266278, doi:10.1016/0003-2697(81)90281-5)
- (en) « Élimination du méthanol dans les Western Blot », sur Université du Michigan (consulté le )
- CP. Moritz, « Tubulin or not tubulin: Heading toward total protein staining as loading control in western blots », Proteomics, (PMID 28941183, DOI 10.1002/pmic.201600189)
- Charlotte Welinder et Lars Ekblad, « Coomassie Staining as Loading Control in Western Blot Analysis », Journal of Proteome Research, vol. 10, no 3, , p. 1416–1419 (ISSN 1535-3893, DOI 10.1021/pr1011476, lire en ligne)
Bibliographie
- Renart J, Reiser J, Stark GR. « Transfer of proteins from gels to diazobenzyloxymethyl-paper and detection with antisera: a method for studying antibody specificity and antigen structure », Proc Natl Acad Sci U S A, 1979 Jul;76(7):3116-20. abstract
- Towbin H, Staehelin T, Gordon J. « Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications », Proc Natl Acad Sci U S A, 1979 Sep;76(9):4350-4. abstract
- Citation's Classic: Burnette
Liens externes
- Explications et mécanismes Elisa et Western Blot avec figures
- (en) Western Blot Protocol
- Principe du western blot Explications très détaillées sur le principe du western blot.
- ASBMB today, january 2012, by Rajendrani Mukhopadhyay, p14-19 http://www.asbmb.org/uploadedFiles/ASBMBToday/Content/Archive/ASBMBToday_2012_01.pdf
- ASBMB today, March 2012, by Rajendrani Mukhopadhyay, p34-35 http://www.asbmb.org/uploadedFiles/ASBMBToday/Content/Archive/ASBMBToday_2012_03.pdf
Voir aussi
- Transfert (biologie moléculaire) pour la liste des techniques de transfert similaires au transfert de protéines.
- Northern blot
- Southern blot
 Portail de la biochimie
Portail de la biochimie