Philadelphia chromosome
| Philadelphia chromosome | |
|---|---|
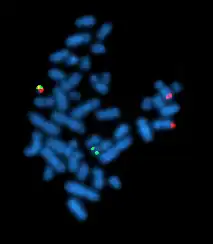 | |
| A metaphase cell positive for the bcr/abl rearrangement using FISH | |
| Specialty | Oncology |
The Philadelphia chromosome or Philadelphia translocation (Ph) is a specific genetic abnormality in chromosome 22 of leukemia cancer cells (particularly chronic myeloid leukemia (CML) cells). This chromosome is defective and unusually short because of reciprocal translocation, t(9;22)(q34;q11), of genetic material between chromosome 9 and chromosome 22, and contains a fusion gene called BCR-ABL1. This gene is the ABL1 gene of chromosome 9 juxtaposed onto the breakpoint cluster region BCR gene of chromosome 22, coding for a hybrid protein: a tyrosine kinase signalling protein that is "always on", causing the cell to divide uncontrollably by interrupting the stability of the genome and impairing various signaling pathways governing the cell cycle.[1]
The presence of this translocation is required for diagnosis of CML; in other words, all cases of CML are positive for BCR-ABL1.[2] (Some cases are confounded by either a cryptic translocation that is invisible on G-banded chromosome preparations, or a variant translocation involving another chromosome or chromosomes as well as the long arm of chromosomes 9 and 22. Other similar but truly Ph-negative conditions are considered CML-like myeloproliferative neoplasms.[3]) However, the presence of the Philadelphia (Ph) chromosome is not sufficiently specific to diagnose CML, since it is also found in acute lymphoblastic leukemia[4] (aka ALL, 25–30% of adult cases and 2–10% of pediatric cases) and occasionally in acute myelogenous leukemia (AML) as well as mixed-phenotype acute leukemia (MPAL).
Molecular biology

The chromosomal defect in the Philadelphia chromosome is a reciprocal translocation, in which parts of two chromosomes, 9 and 22, swap places. The result is that a fusion gene is created by juxtaposing the ABL1 gene on chromosome 9 (region q34) to a part of the BCR (breakpoint cluster region) gene on chromosome 22 (region q11). This is a reciprocal translocation, creating an elongated chromosome 9 (termed a derivative chromosome, or der 9), and a truncated chromosome 22 (the Philadelphia chromosome, 22q-).[5][6] In agreement with the International System for Human Cytogenetic Nomenclature (ISCN), this chromosomal translocation is designated as t(9;22)(q34;q11). The symbol ABL1 is derived from Abelson, the name of a leukemia virus which carries a similar protein. The symbol BCR is derived from breakpoint cluster region, a gene which encodes a protein that acts as a guanine nucleotide exchange factor for Rho GTPase proteins [7]
Translocation results in an oncogenic BCR-ABL1 gene fusion that can be found on the shorter derivative chromosome 22. This gene encodes for a BCR-ABL1 fusion protein. Depending on the precise location of fusion, the molecular weight of this protein can range from 185 to 210 kDa. Consequently, the hybrid BCR-ABL1 fusion protein is referred to as p210 or p185.
Three clinically important variants encoded by the fusion gene are the p190, p210, and p230 isoforms.[8] p190 is generally associated with B-cell acute lymphoblastic leukemia (ALL), while p210 is generally associated with chronic myeloid leukemia but can also be associated with ALL and AML.[9] p230 is usually associated with chronic myelogenous leukemia associated with neutrophilia and thrombocytosis (CML-N).[9] Additionally, the p190 isoform can also be expressed as a splice variant of p210.[10]
The ABL1 gene expresses a membrane-associated protein, a tyrosine kinase, and the BCR-ABL1 transcript is also translated into a tyrosine kinase containing domains from both the BCR and ABL1 genes. The activity of tyrosine kinases is typically regulated in an auto-inhibitory fashion, but the BCR-ABL1 fusion gene codes for a protein that is "always on" or constitutively activated, leading to impaired DNA binding and unregulated cell division (i.e. cancer). This is due to the replacement of the myristoylated cap region, which when present induces a conformational change rendering the kinase domain inactive, with a truncated portion of the BCR protein.[11] Although the BCR region also expresses serine/threonine kinases, the tyrosine kinase function is very relevant for drug therapy. As the N-terminal Y177 and CC domains from BCR encode the constitutive activation of the ABL1 kinase, these regions are targeted in therapies to downregulate BCR-ABL1 kinase activity. Tyrosine kinase inhibitors specific to such domains as CC, Y177, and Rho (such as imatinib and sunitinib) are important drugs against a variety of cancers including CML, renal cell carcinoma (RCC) and gastrointestinal stromal tumors (GISTs).
The fused BCR-ABL1 protein interacts with the interleukin-3 receptor beta(c) subunit and is moderated by an activation loop within its SH1 domain, which is turned “on” when bound to ATP and triggers downstream pathways. The ABL1 tyrosine kinase activity of BCR-ABL1 is elevated relative to wild-type ABL1.[12] Since ABL activates a number of cell cycle-controlling proteins and enzymes, the result of the BCR-ABL1 fusion is to speed up cell division. Moreover, it inhibits DNA repair, causing genomic instability and potentially causing the feared blast crisis in CML.
Proliferative roles in leukemia
The BCR-ABL1 fusion gene and protein encoded by the Philadelphia chromosome affects multiple signaling pathways that directly affect apoptotic potential, cell division rates, and different stages of the cell cycle to achieve unchecked proliferation characteristic of CML and ALL.
JAK/STAT pathway
Particularly vital to the survival and proliferation of myelogenous leukemia cells in the microenvironment of the bone marrow is cytokine and growth factor signaling. The JAK/STAT pathway moderates many of these effectors by activating STATs, which are transcription factors with the ability to modulate cytokine receptors and growth factors. JAK2 phosphorylates the BCR-ABL fusion protein at Y177 and stabilizes the fusion protein, strengthening tumorigenic cell signaling. JAK2 mutations have been shown to be central to myeloproliferative neoplasms and JAK kinases play a central role in driving hematologic malignancies (JAK blood journal). ALL and CML therapies have targeted JAK2 as well as BCR-ABL using nilotinib and ruxolitinib within murine models to downregulate downstream cytokine signaling by silencing STAT3 and STAT5 transcription activation (appelmann et al). The interaction between JAK2 and BCR-ABL within these hematopoietic malignancies implies an important role of JAK-STAT-mediated cytokine signaling in promoting the growth of leukemic cells exhibiting the Ph chromosome and BCR-ABL tyrosine kinase activity. Though the centrality of the JAK2 pathway to direct proliferation in CML has been debated, its role as a downstream effector of the BCR-ABL tyrosine kinase has been maintained. Impacts on the cell cycle via JAK-STAT are largely peripheral, but by directly impacting the maintenance of the hematopoietic niche and its surrounding microenvironment, the BCR-ABL upregulation of JAK-STAT signaling plays an important role in maintaining leukemic cell growth and division.[13][14]
Ras/MAPK/ERK pathway
The Ras/MAPK/ERK pathway relays signals to nuclear transcription factors and plays a role in governing cell cycle control and differentiation. In Ph chromosome-containing cells, the BCR-ABL tyrosine kinase activates the RAS/RAF/MEK/ERK pathway, which results in unregulated cell proliferation via gene transcription in the nucleus. The BCR-ABL tyrosine kinase activates Ras via phosphorylation of the GAB2 protein, which is dependent on BCR-located phosphorylation of Y177. Ras in particular is shown to be an important downstream target of BCR-ABL1 in CML, as Ras mutants in murine models disrupt the development of CML associated with the BCR-ABL1 gene (Effect of Ras inhibition in hematopoiesis and BCR/ABL leukemogenesis). The Ras/RAF/MEK/ERK pathway is also implicated in overexpression of osteopontin (OPN), which is important for maintenance of the hematopoietic stem cell niche, which indirectly influences unchecked proliferation characteristic of leukemic cells. BCR-ABL fusion cells also exhibit constitutively high levels of activated Ras bound to GTP, activating a Ras-dependent signaling pathway which has been shown to inhibit apoptosis downstream of BCR-ABL (Cortez et al). Interactions with the IL-3 receptor also induce the Ras/RAF/MEK/ERK pathway to phosphorylate transcription factors which play a role in driving the G1/S transition of the cell cycle.[15][16][17]
DNA binding and apoptosis
The c-Abl gene in wild-type cells is implicated in DNA binding, which affects such processes as DNA transcription, repair, apoptosis, and other processes underlying the cell cycle. While the nature of this interaction has been debated, evidence exists to suggest that c-Abl phosphorylates HIPK2, a serine/threonine kinase, in response to DNA damage and promotes apoptosis in normal cells. The BCR-ABL fusion, in contrast, has been shown to inhibit apoptosis, but its effect on DNA binding in particular is unclear.[18] In apoptotic inhibition, BCR-ABL cells have been shown to be resistant to drug-induced apoptosis but also have a proapoptotic expression profile by increased expression levels of p53, p21, and Bax. The function of these pro-apoptotic proteins, however, is impaired, and apoptosis is not carried out in these cells. BCR-ABL has also been implicated in preventing caspase 9 and caspase 3 processing, which adds to the inhibitory effect.[19][20][21] Another factor preventing cell cycle progression and apoptosis is the deletion of the IKAROS gene, which presents in >80% of Ph chromosome positive ALL cases. The IKAROS gene is critical to Pre-B cell receptor-mediated cell cycle arrest in ALL cells positive for Ph, which when impaired provides a mechanism for unchecked cell cycle progression and proliferation of defective cells as encouraged by BCR-ABL tyrosine kinase signaling.[22]
Nomenclature
The Philadelphia chromosome is designated Ph (or Ph') chromosome and designates the shortened chromosome 22 which encodes the BCR-ABL fusion gene/protein kinase. It arises from the translocation, which is termed t(9;22)(q34.1;q11.2), between chromosome 9 and chromosome 22, with breaks happening in region (3), band (4), sub-band (1) of the long arm (q) of chromosome 9 and region (1), band (1), sub-band (2) of the long arm (q) of chromosome 22. Hence the chromosome breakpoints are written as (9q34.1) and (22q11.2), respectively, using ISCN standards.
Therapy
Tyrosine kinase inhibitors
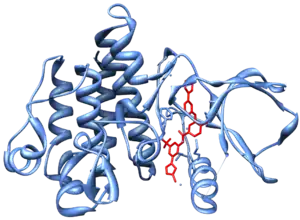
In the late 1990s, STI-571 (imatinib, Gleevec/Glivec) was identified by the pharmaceutical company Novartis (then known as Ciba Geigy) in high-throughput screens for tyrosine kinase inhibitors. Subsequent clinical trials led by Dr. Brian J. Druker at Oregon Health & Science University in collaboration with Dr. Charles Sawyers and Dr. Moshe Talpaz demonstrated that STI-571 inhibits proliferation of BCR-ABL-expressing hematopoietic cells. Although it did not eradicate CML cells, it did greatly limit the growth of the tumor clone and decreased the risk of the feared "blast crisis". In 2000 Dr. John Kuriyan determined the mechanism by which STI-571 inhibits the Abl kinase domain.[23] It was marketed in 2001 by Novartis as imatinib mesylate (Gleevec in the US, Glivec in Europe).
Other pharmacological inhibitors are being developed, which are more potent and/or are active against the emerging Gleevec/Glivec resistant BCR-abl clones in treated patients. The majority of these resistant clones are point-mutations in the kinase of BCR-abl. New inhibitors include dasatinib and nilotinib, which are significantly more potent than imatinib and may overcome resistance. Combination therapies with nilotinib and ruxolitnib have also shown success in suppressing resistance by targeting the JAK-STAT and BCR-ABL stages simultaneously. Small molecule inhibitors, like arsenic trioxide and geldanamycin analogues, have also been identified in downregulating BCR-ABL kinase translation and promoting its degradation by protease.[24][25]
Axitinib, a drug used to treat renal cell carcinoma, has been shown to be effective at inhibiting the Abl kinase activity in patients with BCR-ABL1(T315I).[26] The T315I mutation in the fusion gene confers resistance to other tyrosine kinase inhibitors such as imatinib, however axitinib has been successfully been used to treat a patient with ALL carrying this mutation, as well as CML cells in culture.
Treatment of pediatric Ph+ ALL with a combination of standard chemotherapy and RTK inhibitors may result in remission, but the curative potential is unknown.
Asciminib (Scemblix) was approved for medical use in the United States in October 2021.[27]
Blood or marrow transplants
A potentially curative, but risky, option for pediatric Ph+ ALL or Ph+ CML is bone marrow transplant or cord blood transplant, but chemotherapy is favored by some for achieving first remission (CR1). For some, bone marrow transplant from a matched sibling donor or a matched, unrelated donor may be favored when remission is obtained.
Cord blood transplant is favored by some when a 10/10 bone marrow match is not available, and cord blood transplant may have some advantages, including a reduced incidence of graft-vs-host disease (GVHD), which is a common and significant complication of transplant. However, transplant with cord blood sometimes requires longer periods of time for engraftment, which may increase the potential for complications due to infection. Regardless of the type of transplant, transplant-related mortality and relapse are possible, and the rates may change as treatment protocols improve. For second remission (CR2), if achieved, both chemotherapy and transplant options are possible, and many physicians prefer transplant.
History
The Philadelphia chromosome was first discovered and described in 1959 by David Hungerford at the Lankenau Hospital's Institute for Cancer Research, which merged with the American Oncology Hospital in 1974 to create Fox Chase Cancer Center,[28] along with Peter Nowell from the University of Pennsylvania School of Medicine. The genetic abnormality Hungerford and Nowell found was named after the city in which both organizations were located.[1][29][28][30]
Hungerford was writing his doctoral thesis on chromosomes in a genetics lab at what was then the Institute for Cancer Research at the Lankenau Hospital Research Institute,[28] and detected a flaw in chromosomes from the blood cells of patients with leukemia. This founding observation was the first genetic defect to be linked with a specific human cancer. Nowell was a pathologist at the University of Pennsylvania who was also studying leukemia cells under the microscope when he noticed cells with this genetic flaw in the act of dividing. To his surprise, their chromosomes—usually an indistinct tangle—were visible as separate structures. In searching for an expert on chromosomes, Nowell found Hungerford locally at Lankenau. While conducting his microscopic studies, Hungerford furthered his observations with the discovery that certain leukemia cells had an abnormally short chromosome 22. Subsequently, the mutation he observed became known as the Philadelphia chromosome.
In 1973, Janet Rowley at the University of Chicago identified the mechanism by which the Philadelphia chromosome arises as a translocation.[1][31][32]
See also
References
- 1 2 3 Wapner J (2014). The Philadelphia Chromosome: A Genetic Mystery, a Lethal Cancer, and the Improbable Invention of a Lifesaving Treatment. New York, NY: The Experiment. ISBN 978-1-61519-197-0.
- ↑ "Chronic myeloid leukemia" (PDF). NCCN Clinical Practice Guidelines. National Cancer Comprehensive Network. 15 November 2021. Retrieved 2020-07-15.
- ↑ "Myeloproliferative Neoplasms" (PDF). NCCN Clinical Practice Guidelines. National Cancer Comprehensive Network. 15 November 2021. Retrieved 2018-02-20.
- ↑ Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, et al. (June 2006). "Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias". The New England Journal of Medicine. 354 (24): 2531–2541. doi:10.1056/NEJMoa055229. PMID 16775234.
- ↑ Kurzrock R, Kantarjian HM, Druker BJ, Talpaz M (May 2003). "Philadelphia chromosome-positive leukemias: from basic mechanisms to molecular therapeutics". Annals of Internal Medicine. 138 (10): 819–830. doi:10.7326/0003-4819-138-10-200305200-00010. PMID 12755554. S2CID 25865321.
- ↑ Melo JV (May 1996). "The molecular biology of chronic myeloid leukaemia". Leukemia. 10 (5): 751–756. PMID 8656667.
- ↑ "Gene entry for BCR". NCBI Gene. Retrieved 21 January 2020.
- ↑ Advani AS, Pendergast AM (August 2002). "Bcr-Abl variants: biological and clinical aspects". Leukemia Research. 26 (8): 713–720. doi:10.1016/s0145-2126(01)00197-7. PMID 12191565.
- 1 2 Pakakasama S, Kajanachumpol S, Kanjanapongkul S, Sirachainan N, Meekaewkunchorn A, Ningsanond V, Hongeng S (August 2008). "Simple multiplex RT-PCR for identifying common fusion transcripts in childhood acute leukemia". International Journal of Laboratory Hematology. 30 (4): 286–291. doi:10.1111/j.1751-553X.2007.00954.x. PMID 18665825.
- ↑ Lichty BD, Keating A, Callum J, Yee K, Croxford R, Corpus G, et al. (December 1998). "Expression of p210 and p190 BCR-ABL due to alternative splicing in chronic myelogenous leukaemia". British Journal of Haematology. 103 (3): 711–715. doi:10.1046/j.1365-2141.1998.01033.x. PMID 9858221.
- ↑ Nagar B, Hantschel O, Young MA, Scheffzek K, Veach D, Bornmann W, et al. (March 2003). "Structural basis for the autoinhibition of c-Abl tyrosine kinase". Cell. 112 (6): 859–871. doi:10.1016/s0092-8674(03)00194-6. PMID 12654251.
- ↑ Sattler M, Griffin JD (April 2001). "Mechanisms of transformation by the BCR/ABL oncogene". International Journal of Hematology. 73 (3): 278–291. doi:10.1007/BF02981952. PMID 11345193. S2CID 20999134.
- ↑ Warsch W, Walz C, Sexl V (September 2013). "JAK of all trades: JAK2-STAT5 as novel therapeutic targets in BCR-ABL1+ chronic myeloid leukemia". Blood. 122 (13): 2167–2175. doi:10.1182/blood-2014-04-567289. PMID 23926299.
- ↑ Hantschel O (February 2015). "Targeting BCR-ABL and JAK2 in Ph+ ALL". Blood. 125 (9): 1362–1363. doi:10.1182/blood-2014-12-617548. PMID 25721043.
- ↑ Bandyopadhyay G, Biswas T, Roy KC, Mandal S, Mandal C, Pal BC, et al. (October 2004). "Chlorogenic acid inhibits Bcr-Abl tyrosine kinase and triggers p38 mitogen-activated protein kinase-dependent apoptosis in chronic myelogenous leukemic cells". Blood. 104 (8): 2514–2522. doi:10.1182/blood-2003-11-4065. PMID 15226183.
- ↑ Skorski T, Kanakaraj P, Ku DH, Nieborowska-Skorska M, Canaani E, Zon G, et al. (June 1994). "Negative regulation of p120GAP GTPase promoting activity by p210bcr/abl: implication for RAS-dependent Philadelphia chromosome positive cell growth". The Journal of Experimental Medicine. 179 (6): 1855–1865. doi:10.1084/jem.179.6.1855. PMC 2191514. PMID 8195713.
- ↑ Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA (February 2004). "JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis". Leukemia. 18 (2): 189–218. doi:10.1038/sj.leu.2403241. PMID 14737178.
- ↑ Burke BA, Carroll M (June 2010). "BCR-ABL: a multi-faceted promoter of DNA mutation in chronic myelogeneous leukemia". Leukemia. 24 (6): 1105–1112. doi:10.1038/leu.2010.67. PMC 4425294. PMID 20445577.
- ↑ "The Tyrosine Kinase c-Abl Responds to DNA Damage by Activating the Homeodomain-interacting Protein Kinase 2". Journal of Biological Chemistry. 290 (27): 16489. 2015. doi:10.1074/jbc.p114.628982. PMC 4505403.
- ↑ Kang Z, Liu Y, Xu L, Long Z, Huang D, Yang Y (2011). "Philadelphia Chromosome". Springer Reference. doi:10.1007/springerreference_103128.
- ↑ Kipreos ET, Wang JY (April 1992). "Cell cycle-regulated binding of c-Abl tyrosine kinase to DNA". Science. 256 (5055): 382–385. Bibcode:1992Sci...256..382K. doi:10.1126/science.256.5055.382. PMID 1566087. S2CID 29228735.
- ↑ Qazi S, Uckun FM (December 2013). "Incidence and biological significance of IKZF1/Ikaros gene deletions in pediatric Philadelphia chromosome negative and Philadelphia chromosome positive B-cell precursor acute lymphoblastic leukemia". Haematologica. 98 (12): e151–e152. doi:10.3324/haematol.2013.091140. PMC 3856976. PMID 24323986.
- ↑ Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J (September 2000). "Structural mechanism for STI-571 inhibition of abelson tyrosine kinase". Science. 289 (5486): 1938–1942. Bibcode:2000Sci...289.1938S. doi:10.1126/science.289.5486.1938. PMID 10988075. S2CID 957274.
- ↑ Nimmanapalli R, Bhalla K (December 2002). "Novel targeted therapies for Bcr-Abl positive acute leukemias: beyond STI571". Oncogene. 21 (56): 8584–8590. doi:10.1038/sj.onc.1206086. PMID 12476305.
- ↑ Dan S, Naito M, Tsuruo T (August 1998). "Selective induction of apoptosis in Philadelphia chromosome-positive chronic myelogenous leukemia cells by an inhibitor of BCR - ABL tyrosine kinase, CGP 57148". Cell Death and Differentiation. 5 (8): 710–715. doi:10.1038/sj.cdd.4400400. PMID 10200527.
- ↑ Pemovska T, Johnson E, Kontro M, Repasky GA, Chen J, Wells P, et al. (March 2015). "Axitinib effectively inhibits BCR-ABL1(T315I) with a distinct binding conformation". Nature. 519 (7541): 102–105. Bibcode:2015Natur.519..102P. doi:10.1038/nature14119. PMID 25686603. S2CID 4389086.
- ↑ "FDA approves Novartis Scemblix (asciminib), with novel mechanism of action for the treatment of chronic myeloid leukemia". Novartis (Press release). Retrieved 29 October 2021.
- 1 2 3 "History and Accomplishments". Lankenau Institute for Medical Research.
- ↑ Fox Chase Cancer Center (2015-12-03). "50th Anniversary of the Discovery of the Philadelphia Chromosome".
{{cite journal}}: Cite journal requires|journal=(help) - ↑ "National Academy of Sciences". Science. 132 (3438): 1488–1501. November 1960. Bibcode:1960Sci...132.1488.. doi:10.1126/science.132.3438.1488. PMID 17739576.
- ↑ Rowley JD (June 1973). "Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining". Nature. 243 (5405): 290–293. Bibcode:1973Natur.243..290R. doi:10.1038/243290a0. PMID 4126434. S2CID 26726071.
- ↑ Dreifus C (2011-02-07). "The Matriarch of Modern Cancer Genetics". New York Times.
External links
- Philadelphia+chromosome at the US National Library of Medicine Medical Subject Headings (MeSH)
- bcr-abl+Fusion+Proteins at the US National Library of Medicine Medical Subject Headings (MeSH)