Osteochondrodysplasia
| Osteochondrodysplasia | |
|---|---|
| Other names: Skeletal dysplasia | |
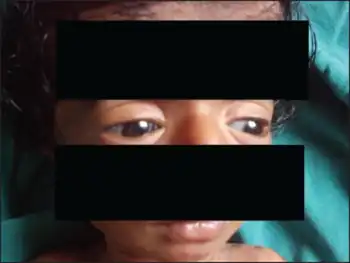 | |
| Characteristic blue sclera in osteogenesis imperfecta | |
| Specialty | Orthopedic |
Osteochondrodysplasia is a general term for a disorder of the development (dysplasia) of bone ("osteo") and cartilage ("chondro").[1] Osteochondrodysplasias are rare diseases. About 1 in 5,000 babies are born with some type of skeletal dysplasia.[2] Nonetheless, if taken collectively, genetic skeletal dysplasias or osteochondrodysplasias comprise a recognizable group of genetically determined disorders with generalized skeletal affection. Osteochondrodysplasias can result in marked functional limitation and even mortality.
Osteochondrodysplasias subtypes can overlap in clinical aspects, therefore plain radiography is absolutely necessary to establish an accurate diagnosis.[3] Magnetic resonance imaging can provide further diagnostic insights and guide treatment strategies especially in cases of spinal involvement. Early diagnosis, and timely management of skeletal dysplasia are important to combat functional deterioration.[3]
Types
Achondroplasia
Achondroplasia is a type of autosomal dominant genetic disorder that is the most common cause of dwarfism. It is also the most common type of non-lethal osteochondrodysplasia or skeletal dysplasia. The prevalence is approximately 1 in 25,000 births.[4] Achondroplastic dwarfs have short stature, with an average adult height of 131 cm (4 feet, 3 inches) for males and 123 cm (4 feet, 0 inches) for females. In achondroplasia the dwarfism is readily apparent at birth. likewise, craniofacial abnormalities in the form of macrocephaly and mid-face hypoplasia are present at birth. The previous clinical findings differentiate between achondroplasia and pseudoachondroplasia in which dwarfism is not recognizable at birth and craniofacial abnormalities are not considered a disease feature. Plain radiography plays an additional and important role in the differential diagnosis of achondroplasia.[3]
Pseudoachondroplasia
Pseudoachondroplasia is an osteochondrodysplasia made distinctive by disproportionate short stature, hip and knee deformities, brachydactyly (short fingers) and ligamentous laxity. It affects at least 1 in 20,000 individuals. Pseudoachondroplasia is inherited in an autosomal dominant manner and is caused solely by mutations in the cartilage oligomeric matrix protein COMP gene.[5] It’s distinguished by a moderate to severe form of disproportionate short-limb short stature. The limb shortening is fundamentally confined to the proximal limb segments i.e. Femurs and humeri. A known presenting feature is a waddling gait, noticed at the onset of walking. A prompt diagnosis of a skeletal dysplasia in general and Pseudoachondroplasia in specific is still based upon a comprehensive clinical and radiographic correlation.[3] A detailed radiographic examination of the axial and appendicular skeleton is invaluable for the differential diagnosis of Pseudoachondroplasia. Coxa vara (reduced neck shaft angle), broad femoral necks, short femurs and humeri, and bullet-shaped vertebrae are noticeable radiographic features. Additionally, the presence of metaphyseal broadening, cupping and dense line of ossification about the knee can simulate rachitic changes. These radiographic features are collectively known as rachitic-like changes. The presence of epiphyseal changes serves as an important differentiating feature from achondroplasia.[3]
Osteogenesis imperfecta
COL1A1/2-related osteogenesis imperfecta is inherited in an autosomal dominant manner. The proportion of cases caused by a De novo COL1A1 or COL1A2 mutations are the cause of osteogenesis imperfecta in the vast majority of perinatally lethal osteogenesis imperfecta, and progressively deforming osteogenesis imperfecta. In classic non-deforming osteogenesis imperfecta with blue sclerae or common variable osteogenesis imperfecta with normal sclerae, nearly 60% of cases are de novo. COL1A1/2-related osteogenesis imperfecta is identified by repeated fractures with trivial trauma, defective dentinogenesis imperfecta (DI), and hearing loss. The clinical features of COL1A1/2-related osteogenesis imperfecta can be highly variable ranging from severe and lethal perinatal fractures to individuals with minimal tendency to repeated fractures and skeletal deformities and with a normal stature and life span. In between the clinical spectrum may include individuals with various degrees of disabling skeletal deformities and short stature.[6] The radiographic findings of osteogenesis imperfecta include; long bone deformations such as bowing of the tibias and femurs, pencil-like deformity and tapering of bones, cortical thinning and rarefaction, pathologic fractures at various degrees of healing, bone shortening and vertebral wedging.[3] Accordingly, COL1A1/2-related osteogenesis imperfecta has been classified into four sub-types (I, II, III, and IV) built upon the diversity of the radioclinical features.[7]
- OI type I: classic non-deforming OI with blue sclerae
- OI type II: perinatally lethal OI
- OI type III: progressively deforming OI
- OI type IV: common variable OI with normal sclerae
Muocopolysachariodosis
Mucopolysaccharidoses (MPS) constitute a commonly seen group of osteochondrodysplasias. Mucopolysaccharidosis can cause a wide spectrum of clinical and radiologic manifestations ranging from mild skeletal and systemic involvement to severe life-threatening manifestations. It is caused by a contiguous gene duplication or deletion syndrome in which multiple genes are involved. All forms of MPS are inherited in an autosomal recessive pattern, except fir of MPS II; Hunter syndrome which is X-linked.[8] They are caused by an abnormal function of the lysosomal enzymes, which blocks degradation of mucopolysaccharides and leads to accumulation of harmful byproducts, namely, heparan sulfate, dermatan sulfate, and keratan sulfate.[9] The resulting cellular malfunction can lead to a diverse array of skeletal and visceral manifestations. MPS have been subcategorized according to the type of enzyme inadequacy and glycoprotein accumulated.[10]
Cleidocranial dysostosis
Cleidocranial dysostosis is a general skeletal condition named for the collarbone (cleido-) and cranium deformities which people with it often have. Common features include:[11]
- Partly or completely missing collarbones.
- A soft spot or larger soft area in the top of the head where the fontanelle failed to close.
- Bones and joints are underdeveloped.
- The permanent teeth include supernumerary teeth.
- Permanent teeth not erupting
- Bossing (bulging) of the forehead.
- Hypertelorism
Fibrous dysplasia
Fibrous dysplasia causes bone thinning[12] and growths or lesions in one or more bones of the human body.
These lesions are tumor-like growths that consist of replacement of the medullary bone with fibrous tissue, causing the expansion and weakening of the areas of bone involved. Especially when involving the skull or facial bones, the lesions can cause externally visible deformities. The skull is often, but not necessarily, affected, and any other bones can be involved.[13]
Langer–Giedion syndrome
Langer–Giedion syndrome is a very rare genetic disorder caused by a deletion of chromosomal material. Diagnosis is usually made at birth or in early childhood. The features associated with this condition include mild to moderate learning difficulties, short stature, unique facial features, small head and skeletal abnormalities including bony growths projecting from the surfaces of bones.[14]
Maffucci syndrome
Maffucci syndrome is a sporadic disease characterized by the presence of multiple enchondromas associated with multiple simple or cavernous soft tissue hemangiomas. Also lymphangiomas may be apparent.[15]
Patients are normal at birth and the syndrome manifests during childhood and puberty. The enchondromas affect the extremities and their distribution is asymmetrical.[16]
Osteosclerosis
Osteosclerosis, an elevation in bone density,[17] is normally detected on an X-ray as an area of whiteness, and is where the bone density has significantly increased. Localized osteosclerosis can be caused by injuries that compress the bone, by osteoarthritis, and osteoma.
Other
- Deformity type Erlenmeyer flask gives a distal femur similar to an Erlenmeyer flask. It may result from Gaucher disease.[18]
- Kashin–Beck disease
- Melnick–Needles syndrome
- Ovine chondrodysplasia
- Familial osteodysplasia, Anderson type
Diagnosis
The diagnosis is mainly based upon delineating the specific clinical and radiographic pattern of skeletal involvement. However, the different types of skeletal dysplasia can overlap considerably in their clinical presentation. Molecular or genetic analysis may be required to resolve diagnostic difficulties. Molecular analysis is also of importance to the phenotype genotype correlations and to disease classifications.
Differential diagnosis
Juvenile idiopathic arthritis may closely resemble the clinical presentation of some osteochondrodysplasias or genetic skeletal dysplsias. In that, both conditions can present with swollen, stiff and deformed joints.[19][20]
Treatment
Emerging therapies for genetic skeletal dysplasias include enzyme replacement therapy,[21] hematopoietic stem cell transplantation[22][23] and gene therapy. These therapies aim at preventing disease progression and thus improving quality of life. Examples of the use of enzyme replacement therapy are Mucopolysaccharidoses[21] and Gaucher disease.[24] Results have shown effectivity of enzyme replacement therapy. In some instances as malignant infantile osteopetrosis, hematopoietic stem cell transplantation can be life-saving.[22][23]
References
- ↑ "Medcyclopaedia - Osteochondrodysplasia". Archived from the original on 2011-05-26.
- ↑ Geister, Krista A.; Camper, Sally A. (2015-01-01). "Advances in Skeletal Dysplasia Genetics". Annual Review of Genomics and Human Genetics. 16 (1): 199–227. doi:10.1146/annurev-genom-090314-045904. PMC 5507692. PMID 25939055.
- 1 2 3 4 5 6 EL-Sobky, TA; Shawky, RM; Sakr, HM; Elsayed, SM; Elsayed, NS; Ragheb, SG; Gamal, R (15 November 2017). "A systematized approach to radiographic assessment of commonly seen genetic bone diseases in children: A pictorial review". J Musculoskelet Surg Res. 1 (2): 25. doi:10.4103/jmsr.jmsr_28_17. S2CID 79825711.
- ↑ Wynn J, King TM, Gambello MJ, Waller DK, Hecht JT (2007). "Mortality in achondroplasia study: A 42-year follow up". Am. J. Med. Genet. A. 143 (21): 2502–11. doi:10.1002/ajmg.a.31919. PMID 17879967. S2CID 25933218.
- ↑ Briggs, MD; Wright, MJ (16 July 2015). "Pseudoachondroplasia". GeneReviews. Archived from the original on 5 December 2020. Retrieved 16 April 2018.
- ↑ Steiner, RD; Adsit, J; Basel, D (14 February 2013). "COL1A1/2 Osteogenesis Imperfecta". COL1A1/2-Related Osteogenesis Imperfecta. GeneReviews. University of Washington, Seattle. Archived from the original on 18 January 2017. Retrieved 16 April 2018.
- ↑ "Osteogenesis Imperfecta - Children's Health Issues". Merck Manuals Consumer Version. Archived from the original on 2022-11-18. Retrieved 2022-11-18.
- ↑ "Mucopolysaccharidoses". NORD (National Organization for Rare Disorders). Archived from the original on 2022-11-18. Retrieved 2022-11-18.
- ↑ "Mucopolysaccharidoses". NORD (National Organization for Rare Disorders). Archived from the original on 2022-11-18. Retrieved 2022-11-18.
- ↑ "Mucopolysaccharidoses - Children's Health Issues". Merck Manuals Consumer Version. Archived from the original on 2022-12-19. Retrieved 2022-11-18.
- ↑ "Cleidocranial Dysplasia". NORD (National Organization for Rare Disorders). Archived from the original on 2016-10-03. Retrieved 2022-11-18.
- ↑ "fibrous dysplasia of bone" at Dorland's Medical Dictionary
- ↑ "Fibrous Dysplasia". NORD (National Organization for Rare Disorders). Archived from the original on 2020-09-20. Retrieved 2022-11-18.
- ↑ Devidayal, null; Marwaha, Ram Kumar (2006-02-01). "Langer-Giedion Syndrome". Indian Pediatrics. 43 (2): 174–175. ISSN 0019-6061. PMID 16528117. Archived from the original on 2022-11-18. Retrieved 2022-11-18 – via PubMed.
- ↑ "Maffucci Syndrome". NORD (National Organization for Rare Disorders). Archived from the original on 2022-12-19. Retrieved 2022-11-18.
- ↑ "Maffucci syndrome: MedlinePlus Genetics". medlineplus.gov. Archived from the original on 2022-11-18. Retrieved 2022-11-18.
- ↑ "Medcyclopaedia - Osteosclerosis". Archived from the original on 2012-02-07. Retrieved 2007-12-23.
- ↑ Marks, Dawn B.; Swanson, Todd; Sandra I Kim; Marc Glucksman (2007). Biochemistry and molecular biology. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins. ISBN 978-0-7817-8624-9.
- ↑ Kaya Akca, U; Simsek Kiper, PO; Urel Demir, G; Sag, E; Atalay, E; Utine, GE; Alikasifoglu, M; Boduroglu, K; Bilginer, Y; Ozen, S (April 2021). "Genetic disorders with symptoms mimicking rheumatologic diseases: A single-center retrospective study". European Journal of Medical Genetics. 64 (4): 104185. doi:10.1016/j.ejmg.2021.104185. PMID 33662637. S2CID 232122235.
- ↑ Elsebaie, H; Mansour, MA; Elsayed, SM; Mahmoud, S; El-Sobky, TA (December 2021). "Multicentric Osteolysis, Nodulosis, and Arthropathy in two unrelated children with matrix metalloproteinase 2 variants: Genetic-skeletal correlations". Bone Reports. 15: 101106. doi:10.1016/j.bonr.2021.101106. PMC 8283316. PMID 34307793.
- 1 2 Jameson, Elisabeth; Jones, Simon; Remmington, Tracey (18 June 2019). "Enzyme replacement therapy with laronidase (Aldurazyme®) for treating mucopolysaccharidosis type I". Cochrane Database of Systematic Reviews. 6: CD009354. doi:10.1002/14651858.CD009354.pub5. PMC 6581069. PMID 31211405.
- 1 2 Hashemi Taheri, Amir Pejman; Radmard, Amir Reza; Kooraki, Soheil; Behfar, Maryam; Pak, Neda; Hamidieh, Amir Ali; Ghavamzadeh, Ardeshir (September 2015). "Radiologic resolution of malignant infantile osteopetrosis skeletal changes following hematopoietic stem cell transplantation: Radiologic Resolution of MIOP After HSCT". Pediatric Blood & Cancer. 62 (9): 1645–1649. doi:10.1002/pbc.25524. PMID 25820806. S2CID 11287381.
- 1 2 El-Sobky, Tamer; El-Haddad, Alaa; Elsobky, Ezzat; Elsayed, Solaf; Sakr, Hossam (1 March 2017). "Reversal of skeletal radiographic pathology in a case of malignant infantile osteopetrosis following hematopoietic stem cell transplantation". The Egyptian Journal of Radiology and Nuclear Medicine. 48 (1): 237–243. doi:10.1016/j.ejrnm.2016.12.013.
- ↑ Shemesh, E; Deroma, L; Bembi, B; Deegan, P; Hollak, C; Weinreb, NJ; Cox, TM (27 March 2015). "Enzyme replacement and substrate reduction therapy for Gaucher disease". The Cochrane Database of Systematic Reviews (3): CD010324. doi:10.1002/14651858.CD010324.pub2. PMID 25812601.
External links
| Classification |
|---|