Chronic recurrent multifocal osteomyelitis
| Chronic recurrent multifocal osteomyelitis | |
|---|---|
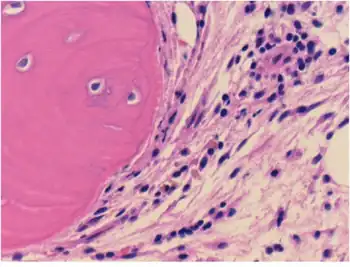 | |
| Connective tissue with mild infiltration by mononuclear inflammatory cells | |
Chronic recurrent multifocal osteomyelitis (CRMO) is a rare condition (1:1,000,000), in which the bones have lesions, inflammation, and pain. It is called multifocal because it can appear in different parts of the body, primarily bones, and osteomyelitis because it is very similar to that disease, although CRMO appears to be without any infection.
The definition of CRMO is evolving. Many doctors and articles described CRMO as an autoimmune disease that has symptoms similar to osteomyelitis, but without the infection. Some doctors thought CRMO was related to SAPHO syndrome. Research now classifies CRMO as an inherited autoinflammatory disease but have yet to isolate the exact gene or other causes responsible for it.
Symptoms and signs
Symptoms may include bone and joint pain, skin redness or inflammation, Inflammatory bowel disease, Psoriasis, and Blister-like lesions on the palms and/or soles of the feet.[1]
Cause
Some specialists believe they have discovered a link between CRMO with a rare allele of marker D18S60, resulting in a haplotype relative risk (HRR) of 18. Other experts found that "mutations in LPIN2 cause a syndromic form of chronic recurrent multifocal osteomyelitis known as Majeed syndrome, while mutations in pstpip2 cause a murine form of the disorder. The roles played by LPIN2 and the human homolog of pstpip2, PSTPIP2, in the cause of chronic recurrent multifocal osteomyelitis are uncertain.[2] The professional theories seem to be moving in the direction of an inherited gene.
Diagnosis
CRMO/CNO is a diagnosis of exclusion. This means that other diseases must be ruled out before the diagnosis can be made. Generally, many tests are required, such as blood tests, x-rays, bone scans, MRI and often a bone biopsy.
Classification
Due to its inflammatory nature, its recurrent flares, and its lack of any known pathogen, CRMO has been reclassified as an autoinflammatory disease. This particular classification encompasses both hereditary types (familial Mediterranean fever, mevalonate kinase deficiency, TNF receptor associated periodic syndrome, cryopyrin-associated periodic syndrome, Blau syndrome, pyogenic sterile arthritis, pyoderma gangrenosum and acne syndrome, CRMO) and multifactorial disorders (Crohn's and Behçet's diseases). CRMO is no longer considered an autoimmune but rather an inherited, autoinflammatory disease.
Treatment
CRMO/CNO is generally treated by a specialist doctor (paediatric rheumatologist) who has experience with patients with CRMO/CNO.
Goals of treatment of CRMO/CNO include:
- Reduce inflammation
- Prevent bone damage and bone deformities
- Decrease pain
CRMO/CNO is different for each patient. Not every child responds to every treatment. Your doctor may need to try several medications before finding the one that works for your child. In severe cases, doctors may combine medications to treat the disease. Your doctor will work with you and your child to help find the best treatment. For some CRMO/CNO patients, the disease can be managed with non-steroidal anti-inflammatory drugs (NSAIDs). NSAIDs are the first line treatment. However, if NSAIDs are not effective, or if your child does not tolerate NSAIDs well, second line treatments are available.
First line treatments include Naproxen (Aleve), Celecoxib (Celebrex) Meloxicam (Mobic), Piroxicam (Feldene), Indomethacin (Indocin), Diclofenac (Voltaren)
Second line treatments include corticosteroids (Prednisone/Prednisolone), Methotrexate (Otrexup, Rasuvo, Trexall), Sulfasalazine (Azulfidine), Pamidronate (Aredia), Zoledronic acid (Zometa), Adalimumab (Humira), Etanercept (Enbrel), Infliximab (Remicade)
These medications are also used in children with other inflammatory and/or bone conditions. Side effects may occur while taking these medications.
Prognosis
Prognosis will depend on your child's individual disease and response to treatment. It is best to discuss the prognosis with your child's pediatric rheumatologist.
Epidemiology
CRMO was once considered strictly a childhood disease, but adults have been diagnosed with it. The affected tends to range from 4 to 14 years old, with 10 as the median age. As stated above, CRMO occurs 1:1,000,000 and primarily in girls with a 5:1 ratio. That means out of six million, there will probably be 5 girls and 1 boy with the condition.
Majeed syndrome
Majeed syndrome is an autoinflammatory disorder consisting of CRMO, congenital dyserythropoietic anemia, and neutrophilic dermatosis. To date, two unrelated families with Majeed syndrome have been reported. Mutations in LPIN2 have been found in both families. Here we report a third consanguineous family with Majeed syndrome with a novel mutation. The patient, a 3-year-old Arabic girl, had hepatosplenomegaly and anemia as a neonate. At age 15 months, she developed recurrent episodes of fever and multifocal osteomyelitis. In addition, bone marrow aspiration demonstrated significant dyserythropoiesis (defective red cell formation), suggesting Majeed syndrome. Coding sequences and splice sites of LPIN2 were sequenced in the patient and her mother. A homozygous single-basepair change was detected in the donor splice site of exon 17 (c.2327+1G>C) in the patient; her mother was heterozygous at this site. These data confirm the role of LPIN2 mutations in the cause of Majeed syndrome.[3]
Congenital dyserythropoietic anemia and chronic recurrent multifocal osteomyelitis, uncommon childhood diseases of unknown cause, occurred in three children (two brothers and a female cousin). Their parents are consanguineous, and the clinical course of their illness was similar. The two brothers also had Sweet syndrome. The association of Sweet syndrome with chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anemia in this family suggests that these rare conditions may be interrelated.[4]
Notes
- ↑ "Chronic recurrent multifocal osteomyelitis | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 2022-01-10. Retrieved 2022-01-10.
- ↑ El-Shanti, HI; Ferguson, PJ (September 2007). "Chronic recurrent multifocal osteomyelitis: a concise review and genetic update". Clinical Orthopaedics and Related Research. 462: 11–9. doi:10.1097/BLO.0b013e3180986d73. PMID 17496555.
- ↑ Al-Mosawi, Al-Saad; Ijadi-Maghsoodi, El-Shanti (2007). "A splice site mutation confirms the role of LPIN2 in Majeed syndrome". Arthritis & Rheumatism. 56 (3): 960–4. doi:10.1002/art.22431. PMID 17330256.
- ↑ Majeed, H; Kalaawi, M; Mohanty, D; Teebi, A; Tunjekar, M; Algharbawy, F; Majeed, S; Algazzar, A (November 1989). "Congenital dyserythropoietic anemia and chronic recurrent multifocal osteomyelitis in three related children and the association with Sweet syndrome in two siblings". The Journal of Pediatrics. 115 (5, Part 1): 730–4. doi:10.1016/S0022-3476(89)80650-X. PMID 2809904.
References
- Brown, Robert; Wilkinson, Timothy (1988). "Chronic recurrent multifocal osteomyelitis". Radiology. 166 (2): 493–6. doi:10.1148/radiology.166.2.3336727. PMID 3336727. Archived from the original on 2008-12-03. Retrieved 2022-01-10.
- Gallagher KT, Roberts RL, MacFarlane JA, Stiehm ER (1997). "Treatment of chronic recurrent multifocal osteomyelitis (CRMO) with interferon-gamma". J. Pediatr. 131 (3): 470–2. doi:10.1016/S0022-3476(97)80081-9. PMID 9329432.
Further reading
- Great Ormond Street Hospital for Children NHS
- GeneReview/NIH/UW entry on Majeed syndrome (Chronic recurrent multifocal osteomyelitis, chronic dyserythropoietic anemia, and transient inflammatory dermatosis) Archived 2022-02-27 at the Wayback Machine
- "A link to many more professional articles and journals." Archived 2022-02-27 at the Wayback Machine
- Chronic recurrent multifocal osteomyelitis at NIH's Office of Rare Diseases
- Majeed syndrome at NIH's Office of Rare Diseases
External links
| Classification | |
|---|---|
| External resources |
|