Contact activation system
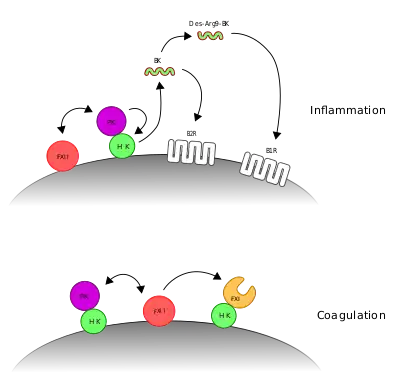
In the contact activation system or CAS, three proteins in the blood, factor XII (FXII), prekallikrein (PK) and high molecular weight kininogen (HK), bind to a surface and cause blood coagulation and inflammation. FXII and PK are proteases and HK is a non-enzymatic co-factor. The CAS can activate the kinin–kallikrein system and blood coagulation through its ability to activate multiple downstream proteins. The CAS is initiated when FXII binds to a surface and reciprocal activation of FXII and PK occurs, forming FXIIa and PKa. FXIIa can initiate the coagulation cascade by cleaving and activating factor XI (FXI), which leads to formation of a blood clot. Additionally, the CAS can activate the kinin–kallikrein system when PKa cleaves HK to form cHK, releasing a peptide known as bradykinin (BK). BK and its derivatives bind to bradykinin receptors B1 and B2 to mediate inflammation.[1][2][3]
Surfaces and activation
Artificial negatively charged substances that activate FXII include L-homocysteine, heparan sulfates, chondroitin sulfates, dermatan sulfate, uric acid crystals, lipoproteins, ferritin and porphyrins. However, the physiological substances or surfaces that activate FXII are still under debate. These may include proteins, such as gC1q-R, aggregated proteins, amyloid, collagen, nucleic acids, and polyphosphates.[4][5][6] The ability of FXII to bind to negatively charged surfaces and activate coagulation forms the basis of the aPTT test, in which artificial materials act as a surface for contact activation. This test is used to measure the contact activation pathway (intrinsic pathway) and the common pathway of clotting.[7] FXII is a zymogen, which means that it requires processing to attain its catalytic protease activity. Upon binding to surfaces, FXII alters in its conformation, giving it low-level protease activity. This change in conformation also promotes its cleavage by PKa and cleavage by FXIIa itself. FXIIa can cleave PK producing PKa, producing a positive feed-back to activate both enzymes. HK binds to PK and is required to locate PK at the surface for activation by FXII.[8]
Zinc has been reportedly demonstrated to be crucial in inducing a conformational change in both FXII and HK as it is required for assembly of FXII and HK bound PK on some negatively charged surfaces. Zinc is suggested to mediate binding of FXII and HK to negatively charged surfaces including gC1q-R and Polyphosphates.[9][10][11]
Contact factors binding to bacteria and viruses
Although contact factors FXII and HK bound PK have been reported to interact with endothelial cells (via gC1q-R), platelets (via Polyphosphate) and Leukocytes; bacteria (Streptococcus pyogenes, Salmonella, and Escherichia coli) and viruses (Hantavirus and Herpes simplex 1 virus) have also been demonstrated to bind to contact factors.[12] Negatively charged lipopolysaccharide (LPS) or surface associated negatively charged teichoic acids from S. aureus[13] and long chain Polyphosphate have all been shown to induce contact activation and bradykinin release thereby contributing to the host-defense reactions[14] by activating the complement cascade.[15]
Physiological roles
Although the contact system can activate FXI and the subsequent clotting cascade, and it is routinely observed to activate coagulation in the presence of medical devices,[16] the actual role of the contact system in normal physiological coagulation remains contentious. This is primarily due to the fact that deficiencies in the contact system proteins FXII, PK and HK do not produce bleeding disorders.[17]
The contact activation system's physiological role in the kinin-kallikrein system is more clear. Here, after activation of PK to PKa by FXIIa, PKa cleaves HK. This produces cleaved HK (cHK), releasing a small peptide known as bradykinin. This peptide binds to bradykinin receptor B2 and its derivative, Des-Arg9-bradykinin binds to bradykinin receptor B1. Upon ligand binding, these receptors mediate inflammatory responses.[18]
Roles in disease
Activation of the CAS is associated with hereditary angioedema, a disorder characterised by episodes of swelling.[19] Genetic knockout studies in murine models of cardiovascular disease and genetic linkage studies in humans have implicated the contact factors in contributing to diverse cardiovascular disease processes including thrombosis[20][21][22] and stroke.[23]
References
- ↑ Schmaier, AH (May 2014). "Physiologic activities of the contact activation system". Thrombosis Research. 133 Suppl 1: S41–4. doi:10.1016/j.thromres.2014.03.018. PMC 4004333. PMID 24759141.
- ↑ de Maat, S; Tersteeg, C; Herczenik, E; Maas, C (June 2014). "Tracking down contact activation - from coagulation in vitro to inflammation in vivo". International Journal of Laboratory Hematology. 36 (3): 374–81. doi:10.1111/ijlh.12222. PMID 24750684.
- ↑ Monika Pathak, Bubacarr Gibril Kaira, Alexandre Slater and Jonas Emsley (2018). "Cell Receptor and Cofactor Interactions of the Contact Activation System and Factor XI". Frontiers in Medicine. 5: 66. doi:10.3389/fmed.2018.00066. PMC 5871670. PMID 29619369.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Ghebrehiwet, B; Kaplan, AP; Joseph, K; Peerschke, EI (November 2016). "The complement and contact activation systems: partnership in pathogenesis beyond angioedema". Immunological Reviews. 274 (1): 281–289. doi:10.1111/imr.12469. PMID 27782339. S2CID 2787614.
- ↑ Naudin, C; Burillo, E; Blankenberg, S; Butler, L; Renné, T (November 2017). "Factor XII Contact Activation". Seminars in Thrombosis and Hemostasis. 43 (8): 814–826. doi:10.1055/s-0036-1598003. PMID 28346966.
- ↑ Schmaier, AH (January 2016). "The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities". Journal of Thrombosis and Haemostasis. 14 (1): 28–39. doi:10.1111/jth.13194. PMID 26565070.
- ↑ Naudin, C; Burillo, E; Blankenberg, S; Butler, L; Renné, T (November 2017). "Factor XII Contact Activation". Seminars in Thrombosis and Hemostasis. 43 (8): 814–826. doi:10.1055/s-0036-1598003. PMID 28346966.
- ↑ de Maat, S; Tersteeg, C; Herczenik, E; Maas, C (June 2014). "Tracking down contact activation - from coagulation in vitro to inflammation in vivo". International Journal of Laboratory Hematology. 36 (3): 374–81. doi:10.1111/ijlh.12222. PMID 24750684.
- ↑ Røjkjær R, Schousboe I (1997). "Partial Identification of the Zn2+‐Binding Sites in Factor XII and its Activation Derivatives". European Journal of Biochemistry. 247 (2): 491–496. doi:10.1111/j.1432-1033.1997.00491.x. PMID 9266689.
- ↑ Schousboe I (1993). "Contact activation in human plasma is triggered by zinc ion modulation of factor XII (Hageman factor)". Blood Coagul Fibrinolysis. 4 (5): 671–8. doi:10.1097/00001721-199310000-00002. PMID 7507361.
- ↑ Rasmus KBJKJER and Inger SCHOUSBOE (1997). "The surface-dependent autoactivation mechanism of factor XII". Eur. J. Biochem. 243 (1–2): 160–166. doi:10.1111/j.1432-1033.1997.0160a.x. PMID 9030735.
- ↑ Mattsson E, Herwald H, Cramer H, Persson K, Sjöbring U, Björck L (2001). "Staphylococcus aureus Induces Release of Bradykinin in Human Plasma". Infection and Immunity. 69 (6): 3877–3882. doi:10.1128/IAI.69.6.3877-3882.2001. PMC 98413. PMID 11349054.
- ↑ Kalter ES, van Dijk WC, Timmerman A, Verhoef J, Bouma BN (1983). "Activation of purified human plasma prekallikrein triggered by cell wall fractions of Escherichia coli and Staphylococcus aureus". The Journal of Infectious Diseases. 148 (4): 682–691. doi:10.1093/infdis/148.4.682. PMID 6355312.
- ↑ Morrissey JH, Choi SH, and Smith SA (2012). "Polyphosphate: an ancient molecule that links platelets, coagulation, and inflammation". Blood. 119 (25): 5972–5979. doi:10.1182/blood-2012-03-306605. PMC 3383012. PMID 22517894.
- ↑ Nickel, KF; Renné, T (2012). "Crosstalk of the plasma contact system with bacteria". Thrombosis Research. 130: S78–S83. doi:10.1016/j.thromres.2012.08.284. PMID 23026673.
- ↑ Jaffer, IH; Fredenburgh, JC; Hirsh, J; Weitz, JI (June 2015). "Medical device-induced thrombosis: what causes it and how can we prevent it?". Journal of Thrombosis and Haemostasis. 13 Suppl 1: S72–81. doi:10.1111/jth.12961. PMID 26149053.
- ↑ de Maat, S; Tersteeg, C; Herczenik, E; Maas, C (June 2014). "Tracking down contact activation - from coagulation in vitro to inflammation in vivo". International Journal of Laboratory Hematology. 36 (3): 374–81. doi:10.1111/ijlh.12222. PMID 24750684.
- ↑ Schmaier, AH (January 2016). "The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities". Journal of Thrombosis and Haemostasis. 14 (1): 28–39. doi:10.1111/jth.13194. PMID 26565070.
- ↑ De Maat, S; Hofman, ZLM; Maas, C (19 June 2018). "Hereditary angioedema: the plasma contact system out of control". Journal of Thrombosis and Haemostasis. 16 (9): 1674–1685. doi:10.1111/jth.14209. PMID 29920929.
- ↑ Fredenburgh JC, Gross PL, and Weitz JI (2017). "Emerging anticoagulant strategies" (PDF). Blood. 129 (2): 147–154. doi:10.1182/blood-2016-09-692996. PMID 27780803.
- ↑ Pierre-Emmanuel Morange; et al. (2011). "KNG1 Ile581Thr and susceptibility to venous thrombosis" (PDF). Blood. 117 (13): 3692–3694. doi:10.1182/blood-2010-11-319053. PMID 21270443.
- ↑ Revenko AS; et al. (2011). "Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding". Blood. 118 (19): 5302–5311. doi:10.1182/blood-2011-05-355248. PMC 4425441. PMID 21821705.
- ↑ Yi Wu (2015). "Contact pathway of coagulation and inflammation". Thrombosis Journal. 13: 17. doi:10.1186/s12959-015-0048-y. PMC 4421925. PMID 25949215.