Delta-beta thalassemia
| Delta-beta thalassemia | |
|---|---|
 | |
| Delta-beta thalassemia | |
| Causes | Produces only gamma-globin and forms HbF(deletes entire delta and beta gene sequence)[1] |
| Diagnostic method | High-performance liquid chromatography |
| Treatment | Blood transfusions[2] |
Delta-beta thalassemia is a rare form of thalassemia in which there is a reduced production of hemoglobin subunit delta and hemoglobin subunit beta and raised levels of hemoglobin subunit gamma. It is an autosomal recessive disorder.[1][3]
Signs and symptoms
An individual with delta-beta thalassemia is usually asymptomatic, however microcytosis can occur where the red blood cells are abnormally small.[1][4]
Mechanism

Delta-beta thalassemia is autosomal recessive disorder,[1] which means both parents are affected and two copies of the gene must be present.[5] A carrier gets a normal gene to produce hemoglobin A, from one parent and the other parent supplies a gene which makes no hemoglobin A.[6] Delta-beta thalassemia is considered rare.[2]
Delta-beta-thalassemia is caused by deletions of the entire delta and beta genes sequences and only gamma-globin and HbF are formed. Rarely, non-deletional forms have been reported. [7][8]
When two delta0 mutations are inherited, no hemoglobin A2 (alpha2, delta2) are formed. This is innocuous because only 2-3% of normal adult hemoglobin is hemoglobin A2. The individual will have normal hematological parameters (erythrocyte count, total hemoglobin, mean corpuscular volume). The delta-beta thalassemia demonstrates one mutation is at the +69 position.[9]
Relation to beta thalassemia
Delta-beta thalassemia can mask the diagnosis of beta thalassemia trait. In beta thalassemia, an increase in hemoglobin A2 results, but the co-existence of a delta-beta thalassemia mutation will decrease the value of the hemoglobin A2 into the normal range, thereby obscuring the diagnosis of beta thalassemia trait[10]
Diagnosis
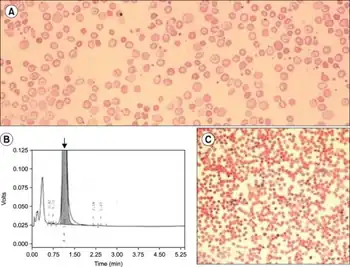
Following the detection of hypochromic microcytic red blood cells, delta-beta thalassemia is confirmed by high-performance liquid chromatography.[11]
Treatment
When needed, treatment for anemia, such as blood transfusions are used.[2]
Stem cell transplant is another option, but the donor and the individual who will receive the bone marrow transplant must be compatible, the risks involved should be evaluated.[2][12][13]
See also
References
- 1 2 3 4 "Delta-beta-thalassemia". Orphanet. Orphanet. Archived from the original on 9 October 2015. Retrieved 16 September 2016.
- 1 2 3 4 "Thalassaemia | Health | Patient". Patient. Archived from the original on 9 September 2016. Retrieved 17 September 2016.
- ↑ "HBD - hemoglobin subunit delta". Orphanet. Orphanet. Archived from the original on 18 September 2016. Retrieved 17 September 2016.
- ↑ Pal, G. K. & (2005). Textbook Of Practical Physiology - 2Nd Edn. Orient Blackswan. p. 53. ISBN 9788125029045. Archived from the original on 18 January 2022. Retrieved 17 September 2016.
- ↑ "Autosomal recessive: MedlinePlus Medical Encyclopedia". medlineplus.gov. Archived from the original on 5 October 2016. Retrieved 17 September 2016.
- ↑ "Delta beta thalassemia carrier" (PDF). Public Health England. Public Health England. Archived from the original (PDF) on 24 September 2016. Retrieved 17 September 2016.
- ↑ "Transcription and Translation - National Human Genome Research Institute (NHGRI)". www.genome.gov. NIH. Archived from the original on 18 September 2016. Retrieved 17 September 2016.
- ↑ Proytcheva, edited by Maria (2010). Diagnostic pediatric hematopathology. Cambridge: Cambridge University Press. p. 61. ISBN 9780521881609. Archived from the original on 18 January 2022. Retrieved 17 September 2016.
- ↑ "OMIM Entry - * 142000 - HEMOGLOBIN--DELTA LOCUS; HBD". www.omim.org. Archived from the original on 30 April 2017. Retrieved 17 September 2016.
- ↑ Galanello, Renzo; Origa, Raffaella (2010). "Beta-thalassemia". Orphanet Journal of Rare Diseases. 5 (1): 11. doi:10.1186/1750-1172-5-11. ISSN 1750-1172. PMC 2893117. PMID 20492708.
- ↑ Ahmad SQ, Zafar SI, Malik HS, Ahmed S (November 2017). "Delta-Beta Thalassaemia in a Pathan Family". Journal of the College of Physicians and Surgeons--Pakistan : JCPSP. 27 (11): 722–724. PMID 29132487.
- ↑ Cao, Antonio; Galanello, Renzo (2010-02-01). "Beta-thalassemia". Genetics in Medicine. 12 (2): 61–76. doi:10.1097/GIM.0b013e3181cd68ed. ISSN 1098-3600. PMID 20098328.
- ↑ "Risks". nhs.uk. Archived from the original on 2018-04-29. Retrieved 2018-04-28.
Further reading
- Verma, S; Bhargava, M; Mittal, SK; Gupta, R (1 January 2013). "Homozygous delta-beta Thalassemia in a Child: a Rare Cause of Elevated Fetal Hemoglobin". Iranian Journal of Pediatric Hematology and Oncology. 3 (1): 222–227. ISSN 2008-8892. PMC 3915439. PMID 24575268.
- Kumar, B. Vinodh; Choccalingam, Chidambharam; Samuel, Premila (1 March 2016). "Incidental Identification of Possible Delta-Beta Thalassemia Trait in a Family: A Rare Cause of Elevated Hb F." Journal of Clinical and Diagnostic Research. 10 (3): BD01–BD02. doi:10.7860/JCDR/2016/16352.7409. ISSN 2249-782X. PMC 4843246. PMID 27134860.
- "Public Health Information Network Vocabulary Access and Distribution System (PHIN VADS)". CDC. Centers for Disease Control. Archived from the original on 18 September 2016. Retrieved 17 September 2016.
External links
| Classification | |
|---|---|
| External resources |
|