Fibrous hamartoma of infancy
| Fibrous hamartoma of infancy | |
|---|---|
| Other names: Subdermal fibromatous tumor of infancy - not recommended[1] | |
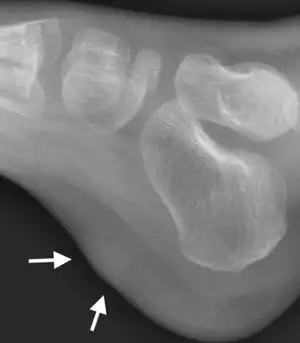 | |
| X-ray foot: Fibrous hamartoma of infancy | |
| Specialty | Dermatology, General surgery, Pathology |
| Symptoms | Benign usually painless tumor in subcutaneous tissues[1] |
| Usual onset | First 2 years of life; congenital in 15%-20% of cases[1] |
| Causes | Unknown[1] |
| Treatment | Surgical resection |
| Prognosis | Excellent, can recur[1] |
| Frequency | Rare, males>females[1] |
Fibrous hamartoma of infancy (FHI) is a non-cancerous soft tissue tumor that develops in very young children.[1] It typically presents as a solitary painless lump just under skin of the axilla, arms, and external genitalia.[1] The overlying skin may be discoloured, swollen, tethered or have excess hair.[1]
It is diagnosed in children who are usually less than 2 years old or, in up to 20% of cases, develops in utero and is diagnosed in an infant at birth.[2][3]
The cells involved in FHI include bland fibroblasts/myofibroblasts, mature fat cells, and primitive-appearing spindle-shaped and/or star-shaped cells.[4] The tumor was first described by R.D. Reye in 1956 who termed the disorder "subdermal fibromatous tumor of infancy" and regarded it as a reactive lesion.[5] In a 1964 study of 30 patients, F.M. Enzinger renamed the tumor hamartoma of infancy; he regarded it to be a hamartoma (i.e. a local malformation of various normal cell types due to a systemic genetic condition) rather than a reactive lesion.[6] However, subsequent studies have found gene mutations in FHI tumor cells and conclude that it is a true neoplasm, i.e. a growth of cells which is uncoordinated with that of the normal surrounding tissue and persists in growing abnormally even if the original trigger is removed.[7] In 2020, the World Health Organization classified FHI in the category of benign fibroblastic and myofibroblastic tumors.[8]
Fibrous hamartoma of infancy is generally a benign tumor but may be locally aggressive,[9] locally infiltration,[10] and in uncommon cases produce symptoms such as tenderness.[11] Surgical excision is the treatment of choice for FHI tumors.[9]
Signs and symptoms
The largest study to date examined 197 cases of FHI. In this study, most individuals presented with a slowly growing, symptomless, subcutaneous mass although rarely these masses were rapidly growing, and/or were tender, painful, warm, and/or were accompanied by skin changes, pigmentation, sweat gland enlargement, and/or increased hair overlaying the tumor. Sixty-eight percent of these cases were diagnosed in the first year of life, 23% were diagnoses at birth, and 9% were diagnosed in children 2-5 years old. The male-to female ratio was 2.4. The tumors were most common in the axilla (23% of cases), upper arm (12.5%), lower arm (10.5%), external genitalia area (9.5%), and inguinal region (7.5%); less common or rare sites for these tumors were the buttocks, back of the head, back of the neck, back of the torso, lower arm, leg, foot, chest, and anal area. Most cases presented as solitary masses but 3 cases presenting with 2 tumors and 1 case presenting with 5 tumors. Overall, the size of these tumors varied between 0.5 to 4.0 cm but in rare cases were as large as 20 cm.[11] Two subsequent studies of 145 and 60 individuals agreed with all of these results but also did diagnose FHI in individuals as old as 8[12] and 14[4] years, respectively.
Genetics
The EGFR gene which codes for the production of the epidermal growth factor receptor protein is located at band 11.2 on the short (or "p") arm of chromosome 7.[13] Studies using next-generation sequencing targeted to the EGFR gene chromosomal area and confirmed by Sanger sequencing have detected insertion and/or deletion mutations in exon 20 of this gene in the tumor cells of 13 of 13 test FHI cases.[14][15] While the activity of the normal EGFR protein is blocked by tyrosine kinase inhibitor drugs, most proteins coded by EGFR exon 20-mutated genes do not respond to these drugs due the structural configuration of their EGFR kinase domains; however, certain protein variants coded by these mutated EGFR genes may still be sensitive, and offer a potential treatment option, for a subset of FHI cases. In any event, the identification of these EGFR can help distinguish FHI from other pediatric spindle cell neoplasms with morphologies similar to FHI such as giant cell fibroblastoma.[7]
Genomic microarray analyses performed on the two FHI cases with tumors that had areas of sarcoma-like morphology found sarcoma-like cells hyperdiploid (i.e. cell with at least 2 more chromosomes than the normal 48), near tetraploid (i.e. four instead of the normal two chromosomes for some but not all chromosomes), single chromosome gains for several chromosomes, and loss of heterozygosity in the p arms of chromosomes 1 and 11 in the first case and in the second case a loss of the p arm in chromosome 10, lose of chromosome 14, and lose of a portion of the q arm of chromosome 22q. These two cases along with recurrent EGFR gene abnormalities strongly support the notation that FHI is as neoplastic tumor.[7]
Pathology
On gross pathological examination, FHI tumors are soft, poorly demarcated, fibro-fatty masses located in subcutaneous tissues.[12] These tumor masses tend to blend with the surrounding fat tissues, may be lobulated, and in rare cases, especially when large, extend into nearby muscles, nerves, and/or fibrous/connective tissue structures.[12] Microscopic histopathological analyses of hematoxylin and eosin stained FHI tumor tissues consistently reveal three very different component zones in virtually all cases: 1) haphazardly arranged, intersecting bundles of bland fibroblast-like/myofibroblast-like cells; 2) mature adipose (i.e. fat) tissue; and 3) highly vascular, myxoid (i.e. connective tissue appearing more blue or purple than normal connective) nodules of primitive-appearing spindled-shaped to star-shaped cells. Chronic inflammatory cell (e.g. lymphocytes, macrophages, and plasma cells) aggregates are commonly present in these lesion. The relative proportion of fibroblast/myofibroblast, adipose tissue, and myxoid zones commonly vary from case to case.[4]
Earlier Immunohistochemical analyses of FHI tumor tissues in a small number of cases have given varying results. A more recent and larger study reported that 75% of tested cases showed variable expression of smooth muscle actin proteins by the cells located primarily in fibroblast/myofibroblast zones and occasionally by the cells located in myxoid zones; the S100 protein was expressed by the fat cells of the adipose tissue zone in all cases; CD34 protein was expressed by the cells in myxoid and fibroblast/myofibroblast zones in all cases; and cells expressing the desmin protein were not detected in any case.[4] A smaller, more recent study also found that 8 of 13 FHI cases had tumor cells in the fibroblast/myofibrobalast zone that expressed smooth-muscle actin protein, 6 of 8 cases had tumor cells in the myxoid zone that expressed the CD34 protein, and 7 of 7 cases had tumor cells in the adipose tissue zone express the S100 protein.[10] The expression pattern of one or more of these proteins has not yet been found to be of help in distinguishing FHI from other tumor types.[16]
Diagnosis
The diagnosis of FHI is dependent on its presentation as a subcutaneous tumor that often occurs in individuals at birth or ages <1-2 years old; its highly characteristic histopathology consisting of fibroblast/myofibroblast, adipose tissue, and myxoid zones; and its content of tumor cells which have one of the EGFR gene mutations described in the previous section.[4][7] The combination of these factors clearly distinguishes FHI from three recently defined tumors (i.e. fibroblastic connective tissue nevus, medallion-like dermal dendrocyte hamartoma, and plaque-like CD34-positive dermal fibroma[16]) as well as various pediatric spindle-shaped cell neoplasms.[7] Tumor imaging methods such as magnetic resonance imaging may also be helpful in suggesting that a tumor is an FHI.[9][17][18]
Treatment
.png.webp)
FHI tumors, if left untreated, have been documented to grow for as long as ~5 years following their diagnosis.[19] The treatment of choice for these tumors is complete surgical excision with clear margins (i.e. with removal of all tumor tissue). Recurrence rates at the site of excision in several studies have been ~15% although rates as low as 1% have been reported by specialized centers.[10] These tumors do not metastasize (i.e. spread to distant tissues) and have an excellent long-term prognosis.[2][9]
See also
References
- 1 2 3 4 5 6 7 8 9 WHO Classification of Tumours Editorial Board, ed. (2020). "1. Soft tissue tumours: Fibrous hamartoma of infancy". Soft Tissue and Bone Tumours: WHO Classification of Tumours. Vol. 3 (5th ed.). Lyon (France): International Agency for Research on Cancer. pp. 59–60. ISBN 978-92-832-4503-2.
- 1 2 Baranov E, Hornick JL (March 2020). "Soft Tissue Special Issue: Fibroblastic and Myofibroblastic Neoplasms of the Head and Neck". Head and Neck Pathology. 14 (1): 43–58. doi:10.1007/s12105-019-01104-3. PMC 7021862. PMID 31950474.
- ↑ Domanski, Henryk A. (2018). "5. Low grade superficial connective tumours". In Klijanienko, Jerzy; Marinšek, Živa Pohar; Domanski, Henryk A. (eds.). Small Volume Biopsy in Pediatric Tumors: An Atlas for Diagnostic Pathology. Springer. p. 219. ISBN 978-3-319-61026-9. Archived from the original on 2023-02-24. Retrieved 2023-02-24.
- 1 2 3 4 5 Al-Ibraheemi A, Martinez A, Weiss SW, Kozakewich HP, Perez-Atayde AR, Tran H, Parham DM, Sukov WR, Fritchie KJ, Folpe AL (April 2017). "Fibrous hamartoma of infancy: a clinicopathologic study of 145 cases, including 2 with sarcomatous features". Modern Pathology. 30 (4): 474–485. doi:10.1038/modpathol.2016.215. PMID 28059097. S2CID 3477224.
- ↑ REYE RD (July 1956). "A consideration of certain subdermal fibromatous tumours of infancy". The Journal of Pathology and Bacteriology. 72 (1): 149–54. doi:10.1002/path.1700720120. PMID 13367990.
- ↑ ENZINGER FM (February 1965). "Fibrous hamartoma of infancy". Cancer. 18 (2): 241–8. doi:10.1002/1097-0142(196502)18:2<241::aid-cncr2820180216>3.0.co;2-c. PMID 14254080.
- 1 2 3 4 5 John I, Fritchie KJ (January 2020). "What is new in pericytomatous, myoid, and myofibroblastic tumors?". Virchows Archiv. 476 (1): 57–64. doi:10.1007/s00428-019-02700-y. PMID 31705190. S2CID 207941071.
- ↑ Sbaraglia M, Bellan E, Dei Tos AP (April 2021). "The 2020 WHO Classification of Soft Tissue Tumours: news and perspectives". Pathologica. 113 (2): 70–84. doi:10.32074/1591-951X-213. PMC 8167394. PMID 33179614.
- 1 2 3 4 Sheybani EF, Eutsler EP, Navarro OM (December 2016). "Fat-containing soft-tissue masses in children". Pediatric Radiology. 46 (13): 1760–1773. doi:10.1007/s00247-016-3690-z. PMID 27866258. S2CID 19576285.
- 1 2 3 Martos-Cabrera L, Sampedro-Ruiz R, Pérez-González YC, Mentzel T, Llamas-Velasco M (June 2021). "Fibrous Hamartoma of Infancy: A Series of 21 Cases and Review of the Literature". Actas Dermo-Sifiliográficas (English Edition). 112 (6): 520–527. doi:10.1016/j.adengl.2021.03.010. PMID 34088477.
- 1 2 Dickey GE, Sotelo-Avila C (1999). "Fibrous hamartoma of infancy: current review". Pediatric and Developmental Pathology. 2 (3): 236–43. doi:10.1007/s100249900119. PMID 10191347. S2CID 3022223.
- 1 2 3 Saab ST, McClain CM, Coffin CM (March 2014). "Fibrous hamartoma of infancy: a clinicopathologic analysis of 60 cases". The American Journal of Surgical Pathology. 38 (3): 394–401. doi:10.1097/PAS.0000000000000104. PMID 24525510. S2CID 40041944.
- ↑ "Archive copy". Archived from the original on 2022-01-23. Retrieved 2021-10-24.
{{cite web}}: CS1 maint: archived copy as title (link) - ↑ Ellington N, Park JY, King K, Josephs S, Rakheja D (August 2017). "EGFR Exon 20 Insertion/Duplication Mutation in Fibrous Hamartoma of Infancy With Predominantly Pseudoangiomatous Pattern Mimicking Giant Cell Fibroblastoma". International Journal of Surgical Pathology. 25 (5): 421–424. doi:10.1177/1066896917698569. PMID 28381148. S2CID 4572547.
- ↑ Park JY, Cohen C, Lopez D, Ramos E, Wagenfuehr J, Rakheja D (December 2016). "EGFR Exon 20 Insertion/Duplication Mutations Characterize Fibrous Hamartoma of Infancy". The American Journal of Surgical Pathology. 40 (12): 1713–1718. doi:10.1097/PAS.0000000000000729. PMID 27631514. S2CID 24148406.
- 1 2 Drabent P, Fraitag S (July 2021). "Update on Superficial Spindle Cell Mesenchymal Tumors in Children". Dermatopathology. 8 (3): 285–300. doi:10.3390/dermatopathology8030035. PMC 8395933. PMID 34449590.
- ↑ Navarro OM (October 2020). "Pearls and Pitfalls in the Imaging of Soft-Tissue Masses in Children". Seminars in Ultrasound, CT, and MR. 41 (5): 498–512. doi:10.1053/j.sult.2020.05.014. PMID 32980096. S2CID 219763567.
- ↑ Ji Y, Hu P, Zhang C, Yan Q, Cheng H, Han M, Huang Z, Wang X, Li H, Han Y (August 2019). "Fibrous hamartoma of infancy: radiologic features and literature review". BMC Musculoskeletal Disorders. 20 (1): 356. doi:10.1186/s12891-019-2743-5. PMC 6679472. PMID 31376836.
- ↑ Xu M, Liu H, Glick S, Khachemoune A (June 2017). "Perianal Lesions in Children: An Updated Review". American Journal of Clinical Dermatology. 18 (3): 343–354. doi:10.1007/s40257-017-0259-z. PMID 28289985. S2CID 3638558.