Pulmonary fibrosis
| Pulmonary fibrosis | |
|---|---|
| Other names: Interstitial pulmonary fibrosis | |
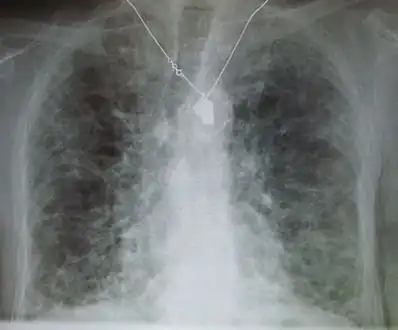 | |
| A chest X-ray demonstrating pulmonary fibrosis believed to be due to amiodarone. | |
| Specialty | Pulmonology |
| Symptoms | Shortness of breath, dry cough, feeling tired, weight loss, nail clubbing[1] |
| Complications | Pulmonary hypertension, respiratory failure, pneumothorax, lung cancer[2] |
| Causes | Environmental pollution, certain medications, connective tissue diseases, interstitial lung disease, unknown[1][3] |
| Treatment | Oxygen therapy, pulmonary rehabilitation, lung transplantation[4] |
| Medication | Pirfenidone, nintedanib[4] |
| Prognosis | Poor[3] |
| Frequency | >5 million people[5] |
Pulmonary fibrosis is a condition in which the lungs become scarred over time.[1] Symptoms include shortness of breath, a dry cough, feeling tired, weight loss, and nail clubbing.[1] Complications may include pulmonary hypertension, respiratory failure, pneumothorax, and lung cancer.[2]
Causes include environmental pollution, certain medications, connective tissue diseases, infections, and interstitial lung diseases.[1][3] Idiopathic pulmonary fibrosis (IPF), an interstitial lung disease of unknown cause, is most common.[1][3] Diagnosis may be based on symptoms, medical imaging, lung biopsy, and lung function tests.[1]
There is no known cure.[1] Treatment is directed towards efforts to improve symptoms and may include oxygen therapy and pulmonary rehabilitation.[1][4] Certain medications may be used to try to slow the worsening of scarring.[4] Lung transplantation may occasionally be an option.[3] At least 5 million people are affected globally.[5] Life expectancy is generally less than five years.[3]
Signs and symptoms

Symptoms of pulmonary fibrosis are mainly:[6]
- Shortness of breath, particularly with exertion
- Chronic dry, hacking coughing
- Fatigue and weakness
- Chest discomfort including chest pain
- Loss of appetite and rapid weight loss
Pulmonary fibrosis is suggested by a history of progressive shortness of breath (dyspnea) with exertion. Sometimes fine inspiratory crackles can be heard at the lung bases on auscultation. A chest X-ray may or may not be abnormal, but high-resolution CT will frequently demonstrate abnormalities.[3]
Cause
Pulmonary fibrosis may be a secondary effect of other diseases. Most of these are classified as interstitial lung diseases. Examples include autoimmune disorders, viral infections and bacterial infection like tuberculosis which may cause fibrotic changes in both lung's upper or lower lobes and other microscopic injuries to the lung. However, pulmonary fibrosis can also appear without any known cause. In this case, it is termed "idiopathic".[7] Most idiopathic cases are diagnosed as idiopathic pulmonary fibrosis. This is a diagnosis of exclusion of a characteristic set of histologic/pathologic features known as usual interstitial pneumonia (UIP). In either case, there is a growing body of evidence which points to a genetic predisposition in a subset of patients. For example, a mutation in surfactant protein C (SP-C) has been found to exist in some families with a history of pulmonary fibrosis.[8] Autosomal dominant mutations in the TERC or TERT genes, which encode telomerase, have been identified in about 15 percent of pulmonary fibrosis patients.[9]
Diseases and conditions that may cause pulmonary fibrosis as a secondary effect include:[3][8]
- Inhalation of environmental and occupational pollutants, such as metals[10] in asbestosis, silicosis and exposure to certain gases. Coal miners, ship workers and sand blasters among others are at higher risk.[7]
- Hypersensitivity pneumonitis, most often resulting from inhaling dust contaminated with bacterial, fungal, or animal products.
- Cigarette smoking can increase the risk or make the illness worse.[7]
- Some typical connective tissue diseases[7] such as rheumatoid arthritis, ankylosing spondylitis, SLE and scleroderma
- Other diseases that involve connective tissue, such as sarcoidosis and granulomatosis with polyangiitis.
- Infections
- Certain medications, e.g. amiodarone, bleomycin (pingyangmycin), busulfan, methotrexate,[7] apomorphine,[11] and nitrofurantoin[12]
- Radiation therapy to the chest
Pathogenesis
Pulmonary fibrosis involves gradual exchange of normal lung parenchyma with fibrotic tissue. The replacement of normal lung with scar tissue causes irreversible decrease in oxygen diffusion capacity, and the resulting stiffness or decreased compliance makes pulmonary fibrosis a restrictive lung disease.[13] Pulmonary fibrosis is perpetuated by aberrant wound healing, rather than chronic inflammation.[14] It is the main cause of restrictive lung disease that is intrinsic to the lung parenchyma. In contrast, quadriplegia[15] and kyphosis[16] are examples of causes of restrictive lung disease that do not necessarily involve pulmonary fibrosis.
Diagnosis
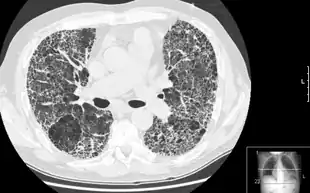
The diagnosis can be confirmed by lung biopsy.[3] A videoscopic assisted thoracoscopic wedge biopsy (VATS) under general anesthesia may be necessary to obtain enough tissue to make an accurate diagnosis. This kind of biopsy involves placement of several tubes through the chest wall, one of which is used to cut off a piece of lung to send for evaluation. The removed tissue is examined histopathologically by microscopy to confirm the presence and pattern of fibrosis as well as presence of other features that may indicate a specific cause e.g. specific types of mineral dust or possible response to therapy e.g. a pattern of so-called non-specific interstitial fibrosis.[3]
Misdiagnosis is common because, while overall pulmonary fibrosis is not rare, each individual type of pulmonary fibrosis is uncommon and the evaluation of patients with these diseases is complex and requires a multidisciplinary approach. Terminology has been standardized but difficulties still exist in their application. Even experts may disagree with the classification of some cases.[17]
On spirometry, as a restrictive lung disease, both the FEV1 (forced expiratory volume in 1 second) and FVC (forced vital capacity) are reduced so the FEV1/FVC ratio is normal or even increased in contrast to obstructive lung disease where this ratio is reduced. The values for residual volume and total lung capacity are generally decreased in restrictive lung disease.[18]
Treatment
Pulmonary fibrosis creates scar tissue. The scarring is permanent once it has developed.[3] Slowing the progression and prevention depends on the underlying cause:
- Treatment options for idiopathic pulmonary fibrosis are very limited. Though research trials are ongoing, there is no evidence that any medications can significantly help this condition. Lung transplantation is the only therapeutic option available in severe cases. Since some types of lung fibrosis can respond to corticosteroids (such as prednisone) and/or other medications that suppress the body's immune system, these types of drugs are sometimes prescribed in an attempt to slow the processes that lead to fibrosis.
- The immune system is felt to play a central role in the development of many forms of pulmonary fibrosis. The goal of treatment with immune suppressive agents such as corticosteroids is to decrease lung inflammation and subsequent scarring. Responses to treatment are variable. Those whose conditions improve with immune suppressive treatment probably do not have idiopathic pulmonary fibrosis, for idiopathic pulmonary fibrosis has no significant treatment or cure.
- Two pharmacological agents intended to prevent scarring in mild idiopathic fibrosis are pirfenidone, which reduced reductions in the 1-year rate of decline in FVC. Pirfenidone also reduced the decline in distances on the 6-minute walk test, but had no effect on respiratory symptoms.[19] The second agent is nintedanib, which acts as antifibrotic, mediated through the inhibition of a variety of tyrosine kinase receptors (including platelet-derived growth factor, fibroblast growth factor, and vascular endothelial growth factor).[20] A randomized clinical trial showed it reduced lung-function decline and acute exacerbations.[21]
- Anti-inflammatory agents have only limited success in reducing the fibrotic process. Some of the other types of fibrosis, such as non-specific interstitial pneumonia, may respond to immunosuppressive therapy such as corticosteroids. However, only a minority of patients respond to corticosteroids alone, so additional immunosuppressants, such as cyclophosphamide, azathioprine, methotrexate, penicillamine, and cyclosporine may be used. Colchicine has also been used with limited success.[3] There are ongoing trials with newer drugs such as IFN-γ and mycophenolate mofetil.
- Hypersensitivity pneumonitis, a less severe form of pulmonary fibrosis, is prevented from becoming aggravated by avoiding contact with the causative material.
- Oxygen supplementation improves the quality of life and exercise capacity. Lung transplantation may be considered for some patients.[22]
Prognosis
.jpg.webp)
Hypoxia caused by pulmonary fibrosis can lead to pulmonary hypertension, which, in turn, can lead to heart failure of the right ventricle. Hypoxia can be prevented with oxygen supplementation.[3]
Pulmonary fibrosis may also result in an increased risk for pulmonary emboli, which can be prevented by anticoagulants.[3]
Epidemiology
Five million people worldwide are affected by pulmonary fibrosis. The rates below are per 100,000 persons, and the ranges reflect narrow and broad inclusion criteria, respectively.
| Incidence rate | Prevalence rate | Population | Years covered | Reference |
|---|---|---|---|---|
| 6.8-16.3 | 14.0-42.7 | U.S. health care claims processing system | 1996–2000 | Raghu et al. [23] |
| 8.8-17.4 | 27.9-63.0 | Olmsted County, MN | 1997–2005 | Fernandez Perez et al. [24] |
| 27.5 | 30.3 | Males in Bernalillo County, NM | 1988–1990 | Coultas et al. [25] |
| 11.5 | 14.5 | Females |
Based on these rates, pulmonary fibrosis prevalence in the United States could range from more than 29,000 to almost 132,000, based on the population in 2000 that was 18 years or older. The actual numbers may be significantly higher due to misdiagnosis. Typically, patients are in their forties and fifties when diagnosed while the incidence of idiopathic pulmonary fibrosis increases dramatically after the age of fifty. However, loss of pulmonary function is commonly ascribed to old age, heart disease or to more common lung diseases.
References
- 1 2 3 4 5 6 7 8 9 "Pulmonary Fibrosis". medlineplus.gov. Archived from the original on 5 July 2016. Retrieved 20 December 2019.
- 1 2 "Pulmonary fibrosis - Symptoms and causes". Mayo Clinic. Archived from the original on 15 July 2014. Retrieved 20 December 2019.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 "Pulmonary Fibrosis". MedicineNet, Inc. Archived from the original on 19 July 2014. Retrieved 26 July 2014.
- 1 2 3 4 "Pulmonary fibrosis - Diagnosis and treatment - Mayo Clinic". www.mayoclinic.org. Archived from the original on 4 July 2014. Retrieved 20 December 2019.
- 1 2 "American Thoracic Society - General Information about Pulmonary Fibrosis". www.thoracic.org. Archived from the original on 20 December 2019. Retrieved 20 December 2019.
- ↑ Mayo Clinic Staff. "Symptoms". Mayo Foundation for Medical Education and Research. Archived from the original on 4 July 2014. Retrieved 26 July 2014.
- 1 2 3 4 5 MedlinePlus > Pulmonary Fibrosis Archived 5 July 2016 at the Wayback Machine Date last updated: 9 February 2010
- 1 2 Mayo Clinic Staff. "Causes". Mayo Foundation for Medical Education and Research. Archived from the original on 1 October 2014. Retrieved 26 July 2014.
- ↑ "Idiopathic pulmonary fibrosis". Genetics Home Reference, United States National Library of Medicine. Archived from the original on 26 September 2018. Retrieved 17 August 2018.
- ↑ Hubbard R, Cooper M, Antoniak M, et al. (2000). "Risk of cryptogenic fibrosing alveolitis in metal workers". Lancet. 355 (9202): 466–467. doi:10.1016/S0140-6736(00)82017-6. PMID 10841131.
- ↑ "Not Found - BIDMC". bidmc.org. Archived from the original on 16 March 2014. Retrieved 29 April 2018.
- ↑ Goemaere NN, Grijm K, van Hal PT, den Bakker MA (2008). "Nitrofurantoin-induced pulmonary fibrosis: a case report". J Med Case Rep. 2: 169. doi:10.1186/1752-1947-2-169. PMC 2408600. PMID 18495029.
- ↑ Mayo Clinic Staff. "Complications". Mayo Foundation for Medical Education and Research. Archived from the original on 4 July 2014. Retrieved 26 July 2014.
- ↑ Gross TJ, Hunninghake GW (2001). "Idiopathic pulmonary fibrosis". N Engl J Med. 345 (7): 517–525. doi:10.1056/NEJMra003200. PMC 2231521. PMID 16928146.
- ↑ Walker J, Cooney M, Norton S (August 1989). "Improved pulmonary function in chronic quadriplegics after pulmonary therapy and arm ergometry". Paraplegia. 27 (4): 278–83. doi:10.1038/sc.1989.42. PMID 2780083.
- ↑ eMedicine Specialties > Pulmonology > Interstitial Lung Diseases > Restrictive Lung Disease Archived 5 March 2010 at the Wayback Machine Author: Lalit K Kanaparthi, MD, Klaus-Dieter Lessnau, MD, Sat Sharma, MD. Updated: 27 July 2009
- ↑ Mayo Clinic Staff. "Tests and diagnosis". Mayo Foundation for Medical Education and Research. Archived from the original on 4 July 2014. Retrieved 26 July 2014.
- ↑ "www.spirXpert.com". Archived from the original on 28 January 2010.
- ↑ King TE Jr; Bradford WZ; Castro-Bernardini S; et al. (May 2014). "A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis" (PDF). NEJM. 370 (22): 2083–2092. doi:10.1056/NEJMoa1402582. PMID 24836312. Archived (PDF) from the original on 19 December 2021. Retrieved 2 July 2019.
- ↑ Richeldi L, Costabel U, Selman M, et al. (2011). "Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis". N Engl J Med. 365 (12): 1079–1087. doi:10.1056/nejmoa1103690. PMID 21992121.
- ↑ Richeldi L, du Bois RM, Raghu G, et al. (May 2014). "Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis" (PDF). N Engl J Med. 370 (22): 2071–2082. doi:10.1056/NEJMoa1402584. hdl:11365/974374. PMID 24836310. Archived (PDF) from the original on 28 August 2021. Retrieved 3 September 2019.
- ↑ Mayo Clinic Staff. "Lifestyle and home remedies". Mayo Foundation for Medical Education and Research. Archived from the original on 4 July 2014. Retrieved 26 July 2014.
- ↑ Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and Prevalence of Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2006;174:810-6.
- ↑ Fernandez Perez ER, Daniels CE, Schroeder DR, St Sauver J, Hartman TE, Bartholmai BJ, Yi ES, Ryu JH. Incidence, Prevalence, and Clinical Course of Idiopathic Pulmonary Fibrosis: A Population-Based Study. Chest. Jan 2010;137:129-37.
- ↑ Coultas DB, Zumwalt RE, Black WC, Sobonya RE. The Epidemiology of Interstitial Lung Diseases. Am J Respir Crit Care Med. Oct 1994;150(4):967-72. cited by Michaelson JE, Aguayo SM, Roman J. Idiopathic Pulmonary Fibrosis: A Practical Approach for Diagnosis and Management. Chest. Sept 2000;118:788-94.
External links
| Classification |
|---|