Renal–hepatic–pancreatic dysplasia
| Renal–hepatic–pancreatic dysplasia | |
|---|---|
| Other names: Ivemark II syndrome, Renohepaticopancreatic dysplasia | |
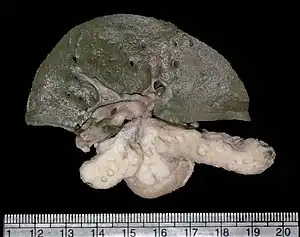 | |
| Gross photo of liver and pancreas showing multiple cysts in the latter in a patient with renal–hepatic–pancreatic dysplasia | |
Renal–hepatic–pancreatic dysplasia is an autosomal recessive congenital disorder characterized by pancreatic fibrosis, renal dysplasia and hepatic dysgenesis. It is usually fatal soon after birth.An association with NPHP3 has been described.[1] It was characterized in 1959.[2][3]
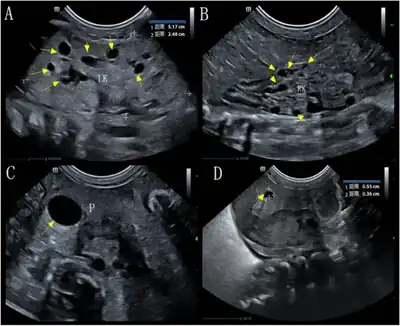
a,b)Image upper abdomen showed left & right kidney multiple cysts c,d) pancreas contained two cysts
References
- ↑ Bergmann C, Fliegauf M, Brüchle NO, et al. (April 2008). "Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia". Am. J. Hum. Genet. 82 (4): 959–970. doi:10.1016/j.ajhg.2008.02.017. PMC 2427297. PMID 18371931.
- ↑ Vankalakunti M, Gupta K, Kakkar N, Das A (2007). "Renal-hepatic-pancreatic dysplasia syndrome (Ivemark's syndrome)". Diagn Pathol. 2: 24. doi:10.1186/1746-1596-2-24. PMC 1919354. PMID 17605805.
- ↑ IVEMARK BI, OLDFELT V, ZETTERSTROM R (January 1959). "Familial dysplasia of kidneys, liver and pancreas: a probably genetically determined syndrome". Acta Paediatr. 48 (1): 1–11. doi:10.1111/j.1651-2227.1959.tb16011.x. PMID 13626573. S2CID 196353197.
External links
- GeneReview/NCBI/NIH/UW entry on Refsum Disease Archived 2010-05-28 at the Wayback Machine
| Classification | |
|---|---|
| External resources |
|
This article is issued from Offline. The text is licensed under Creative Commons - Attribution - Sharealike. Additional terms may apply for the media files.