Severe combined immunodeficiency
| Severe Combined Immune Deficiency | |
|---|---|
| Other names: Alymphocytosis, Glanzmann–Riniker syndrome, Severe mixed immunodeficiency syndrome, and Thymic alymphoplasia[1] | |
 | |
| David Vetter, a child born in 1971 with severe combined immunodeficiency (SCID). | |
| Treatment | Bone marrow transplantation and prophylaxis against infection |
| Medication | IVIG, gene therapy |
| Frequency | 1 in 50,000 to 100,000 (X-linked form) |
Severe combined immunodeficiency (SCID), also known as Swiss-type agammaglobulinemia, is a rare genetic disorder characterized by the disturbed development of functional T cells and B cells caused by numerous genetic mutations that result in differing clinical presentations.[2] SCID involves defective antibody response due to either direct involvement with B lymphocytes or through improper B lymphocyte activation due to non-functional T-helper cells.[3] Consequently, both "arms" (B cells and T cells) of the adaptive immune system are impaired due to a defect in one of several possible genes. SCID is the most severe form of primary immunodeficiencies,[4] and there are now at least nine different known genes in which mutations lead to a form of SCID.[5] It is also known as the bubble boy disease and bubble baby disease because its victims are extremely vulnerable to infectious diseases and some of them, such as David Vetter, have become famous for living in a sterile environment. SCID is the result of an immune system so highly compromised that it is considered almost absent.
SCID patients are usually affected by severe bacterial, viral, or fungal infections early in life and often present with interstitial lung disease, chronic diarrhea, and failure to thrive.[3] Ear infections, recurrent Pneumocystis jirovecii (previously carinii) pneumonia, and profuse oral candidiasis commonly occur. These babies, if untreated, usually die within one year due to severe, recurrent infections unless they have undergone successful hematopoietic stem cell transplantation or gene therapy in clinical trials.[6]
Classification
| Type | Description |
|---|---|
| X-linked severe combined immunodeficiency | Most cases of SCID are due to mutations in the IL2RG gene encoding the common gamma chain (γc) (CD132), a protein that is shared by the receptors for interleukins IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21. These interleukins and their receptors are involved in the development and differentiation of T and B cells. Because the common gamma chain is shared by many interleukin receptors, mutations that result in a non-functional common gamma chain cause widespread defects in interleukin signalling. The result is a near complete failure of the immune system to develop and function, with low or absent T cells and NK cells and non-functional B cells. The common gamma chain is encoded by the gene IL-2 receptor gamma, or IL-2Rγ, which is located on the X-chromosome. For this reason, immunodeficiency caused by mutations in IL-2Rγ is known as X-linked severe combined immunodeficiency. The condition is inherited in an X-linked recessive pattern. |
| Adenosine deaminase deficiency | The second most common form of SCID after X-SCID is caused by a defective enzyme, adenosine deaminase (ADA), necessary for the breakdown of purines. Lack of ADA causes accumulation of dATP. This metabolite will inhibit the activity of ribonucleotide reductase, the enzyme that reduces ribonucleotides to generate deoxyribonucleotides. The effectiveness of the immune system depends upon lymphocyte proliferation and hence dNTP synthesis. Without functional ribonucleotide reductase, lymphocyte proliferation is inhibited and the immune system is compromised. |
| Purine nucleoside phosphorylase deficiency | An autosomal recessive disorder involving mutations of the purine nucleoside phosphorylase (PNP) gene. PNP is a key enzyme in the purine salvage pathway. Impairment of this enzyme causes elevated dGTP levels resulting in T-cell toxicity and deficiency. |
| Reticular dysgenesis | Inability of granulocyte precursors to form granules secondary to mitochondrial adenylate kinase 2 (AK2) malfunction. |
| Omenn syndrome | The manufacture of immunoglobulins requires recombinase enzymes derived from the recombination activating genes RAG-1 and RAG-2. These enzymes are involved in the first stage of V(D)J recombination, the process by which segments of a B cell or T cell's DNA are rearranged to create a new T cell receptor or B cell receptor (and, in the B cell's case, the template for antibodies). Certain mutations of the RAG-1 or RAG-2 genes prevent V(D)J recombination, causing SCID.[7] |
| Bare lymphocyte syndrome | Type 1: MHC class I is not expressed on the cell surface. The defect is caused by defective TAP proteins, not the MHC-I protein.
Type 2: MHC class II is not expressed on the cell surface of all antigen presenting cells. Autosomal recessive. The MHC-II gene regulatory proteins are what is altered, not the MHC-II protein itself. |
| JAK3 | Janus kinase-3 (JAK3) is an enzyme that mediates transduction downstream of the γc signal. Mutation of its gene causes SCID.[8] |
| DCLRE1C | DCLRE1C "Artemis" is a gene required for DNA repair and V(D)J recombination. A recessive loss-of-function mutation found the Navajo and Apache population causes SCID and radiation intolerance.[9][10] |
Signs and symptoms
The clinical presentation of this condition is consistent with, but not limited to the following:[11]
- Failure to thrive
- Fever
- Skin rash
- Frequent respiratory infections
- Infection in blood stream
Genetics
In terms of the inheritance of this condition , it can be either X-linked recessive or autosomal recessive manner although X-linked SCID is the most common type of SCID [11]
Diagnosis
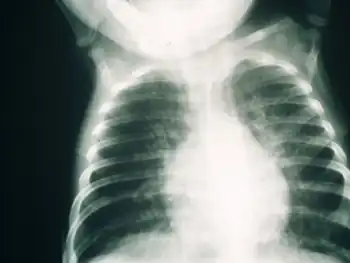
Early diagnosis of SCID is usually difficult due to the need for advanced screening techniques. Several symptoms may indicate a possibility of SCID in a child, such as a family history of infant death, chronic coughs, hyperinflated lungs, and persistent infections. A full blood lymphocyte count is often considered a reliable manner of diagnosing SCID, but higher lymphocyte counts in childhood may influence results. Clinical diagnosis based on genetic defects is also a possible diagnostic procedure that has been implemented in the UK.[12]
Some SCID can be detected by sequencing fetal DNA if a known history of the disease exists. Otherwise, SCID is not diagnosed until about six months of age, usually indicated by recurrent infections. The delay in detection is because newborns carry their mother's antibodies for the first few weeks of life and SCID babies look normal.
Newborn screening
Several countries test all newborns for SCID as a part of routine newborn screening. All states in the U.S.[13] are performing screening for SCID in newborns using real-time quantitative PCR to measure the concentration of T-cell receptor excision circles.[14] United Kingdom intends to introduce newborn screening for SCID in September 2021.[15]
Treatment
The most common treatment for SCID is bone marrow transplantation, which has been very successful using either a matched related or unrelated donor, or a half-matched donor, who would be either parent. The half-matched type of transplant is called haploidentical. Haploidentical bone marrow transplants require the donor marrow to be depleted of all mature T cells to avoid the occurrence of graft-versus-host disease (GVHD).[16] Consequently, a functional immune system takes longer to develop in a patient who receives a haploidentical bone marrow transplant compared to a patient receiving a matched transplant. The first reported case of successful transplant was a Spanish child patient who was interned in Memorial Sloan Kettering Cancer Center in 1982, in New York City.[16] David Vetter, the original "bubble boy", had one of the first transplantations also, but eventually died because of an unscreened virus, Epstein-Barr (tests were not available at the time), in his newly transplanted bone marrow from his sister, an unmatched bone marrow donor. Today, transplants done in the first three months of life have a high success rate. Physicians have also had some success with in utero transplants done before the child is born and also by using cord blood which is rich in stem cells. In utero transplants allow for the fetus to develop a functional immune system in the sterile environment of the uterus;[17] however complications such as GVHD would be difficult to detect or treat if they were to occur.[18]
More recently gene therapy has been attempted as an alternative to the bone marrow transplant. Transduction of the missing gene to hematopoietic stem cells using viral vectors is being tested in ADA SCID and X-linked SCID. In 1990, four-year-old Ashanthi DeSilva became the first patient to undergo successful gene therapy. Researchers collected samples of DeSilva's blood, isolated some of her white blood cells, and used a retrovirus to insert a healthy adenosine deaminase (ADA) gene into them. These cells were then injected back into her body, and began to express a normal enzyme. This, augmented by weekly injections of ADA, corrected her deficiency. However, the concurrent treatment of ADA injections may impair the success of gene therapy, since transduced cells will have no selective advantage to proliferate if untransduced cells can survive in the presence of the injected ADA.[19]
In 2000, a gene therapy "success" resulted in SCID patients with a functional immune system. These trials were stopped when it was discovered that two of ten patients in one trial had developed leukemia resulting from the insertion of the gene-carrying retrovirus near an oncogene. In 2007, four of the ten patients have developed leukemias.[20] Work aimed at improving gene therapy is now focusing on modifying the viral vector to reduce the likelihood of oncogenesis and using zinc-finger nucleases to further target gene insertion.[21] No leukemia cases have yet been seen in trials of ADA-SCID, which does not involve the gamma c gene that may be oncogenic when expressed by a retrovirus.
From the treatments of Ashanthi DeSilva in 1990 which is considered gene therapy's first success until 2014 around 60 patients were treated for either ADA-SCID or X-SCID[22] using retroviruses vectors but as previously mentioned the occurrence of cases developing leukemia forced to make changes to improve safety,[23] more recently in 2019 a new method using an altered version of the HIV virus as a lentivirus vector was reported in the treatment of 8 children with X-SCID,[24][25][26][6] and in 2021 the same method was used in 50 children with ADA-SCID obtaining positive results in 48 of them.[27][28][29]
There are also some non-curative methods for treating SCID. Reverse isolation involves the use of laminar air flow and mechanical barriers (to avoid physical contact with others) to isolate the patient from any harmful pathogens present in the external environment.[30] A non-curative treatment for patients with ADA-SCID is enzyme replacement therapy, in which the patient is injected with polyethyleneglycol-coupled adenosine deaminase (PEG-ADA) which metabolizes the toxic substrates of the ADA enzyme and prevents their accumulation.[19] Treatment with PEG-ADA may be used to restore T cell function in the short term, enough to clear any existing infections before proceeding with curative treatment such as a bone marrow transplant.[31]
Epidemiology
The most commonly quoted figure for the prevalence of SCID is around 1 in 100,000 births, although this is regarded by some to be an underestimate of the true prevalence;[32] some estimates predict that the prevalence rate is as high as 1 in 50,000 live births.[3] A figure of about 1 in 65,000 live births has been reported for Australia.[33]
Due to the particular genetic nature of SCID, a higher prevalence may be found in certain regions and associated cultures where higher rates of consanguineous mating occur.[34] A Moroccan study reported that consanguineous parenting was observed in 75% of the families of Moroccan SCID patients.[35]
Recent studies indicate that one in every 2,500 children in the Navajo population inherit severe combined immunodeficiency. This condition is a significant cause of illness and death among Navajo children.[9] Ongoing research reveals a similar genetic pattern among the related Apache people.[10]
SCID in animals
SCID mice were and still are used in disease, vaccine, and transplant research; especially as animal models for testing the safety of new vaccines or therapeutic agents in people with weakened immune system.
Recessive gene with clinical signs similar to the human condition, also affects the Arabian horse. In horses, the condition remains a fatal disease, as the animal inevitably succumbs to an opportunistic infection within the first four to six months of life.[36] However, carriers, who themselves are not affected by the disease, can be detected with a DNA test. Thus careful breeding practices can avoid the risk of an affected foal being produced.[37]
Another animal with well-characterized SCID pathology is the dog. There are two known forms, an X-linked SCID in Basset Hounds that has similar ontology to X-SCID in humans[38] and an autosomal recessive form seen in one line of Jack Russell Terriers that is similar to SCID in Arabian horses and mice.[39]
SCID mice also serve as a useful animal model in the study of the human immune system and its interactions with disease, infections, and cancer.[40] For example, normal strains of mice can be lethally irradiated, killing all rapidly dividing cells. These mice then receive bone marrow transplantation from SCID donors, allowing engraftment of human peripheral blood mononuclear cells (PBMC) to occur. This method can be used to study whether T cell-lacking mice can perform hematopoiesis after receiving human PBMC.[41]
See also
- David Vetter
- Aisha Chaudhary
- List of cutaneous conditions
- List of radiographic findings associated with cutaneous conditions
References
- ↑ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 978-1-4160-2999-1.
- ↑ Burg M, Gennery AR (2011). "Educational paper: The expanding clinical and immunological spectrum of severe combined immunodeficiency". Eur J Pediatr. 170 (5): 561–571. doi:10.1007/s00431-011-1452-3. PMC 3078321. PMID 21479529.
- 1 2 3 Aloj G, Giardano G, Valentino L, Maio F, Gallo V, Esposito T, Naddei R, Cirillo E, Pignata C (2012). "Severe combined immunodeficiencies: New and Old Scenarios". Int Rev Immunol. 31 (1): 43–65. doi:10.3109/08830185.2011.644607. PMID 22251007. S2CID 24088244.
- ↑ Cavazanna-Calvo M, Hacein-Bey S, Yates F, de Villartay JP, Le Deist F, Fischer A (2001). "Gene therapy of severe combined immunodeficiencies". J Gene Med. 3 (3): 201–206. doi:10.1002/1521-2254(200105/06)3:3<201::AID-JGM195>3.0.CO;2-Z. PMID 11437325.
- ↑ Buckley R (2003). "Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution". Annu Rev Immunol. 22: 625–655. doi:10.1146/annurev.immunol.22.012703.104614. PMID 15032591.
- 1 2 Rohr, Karen (2019-04-17). "Gene therapy restores immunity in infants with rare immunodeficiency disease". National Institutes of Health (NIH). Archived from the original on 2020-06-04. Retrieved 2020-06-04.
- ↑ Haq IJ, Steinberg LJ, Hoenig M, et al. (2007). "GvHD-associated cytokine polymorphisms do not associate with Omenn syndrome rather than T-B- SCID in patients with defects in RAG genes". Clin. Immunol. 124 (2): 165–9. doi:10.1016/j.clim.2007.04.013. PMID 17572155.
- ↑ Pesu M, Candotti F, Husa M, Hofmann SR, Notarangelo LD, O'Shea JJ (2005). "Jak3, severe combined immunodeficiency, and a new class of immunosuppressive drugs". Immunol. Rev. 203: 127–42. doi:10.1111/j.0105-2896.2005.00220.x. PMID 15661026. S2CID 20684919. Archived from the original on 2021-10-08. Retrieved 2022-01-14.
- 1 2 Li L, Moshous D, Zhou Y, et al. (2002). "A founder mutation in Artemis, an SNM1-like protein, causes SCID in Athabascan-speaking Native Americans". J. Immunol. 168 (12): 6323–9. doi:10.4049/jimmunol.168.12.6323. PMID 12055248.
- 1 2 "Severe combined immunodeficiency | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 21 December 2021. Retrieved 6 February 2022.
- ↑ Gennery, A; Cant, A (March 2001). "Diagnosis of severe combined immunodeficiency". J Clin Pathol. 54 (3): 191–195. doi:10.1136/jcp.54.3.191. PMC 1731376. PMID 11253129.
- ↑ van der Burg, Mirjam; Mahlaoui, Nizar; Gaspar, Hubert Bobby; Pai, Sung-Yun (2019-09-18). "Universal Newborn Screening for Severe Combined Immunodeficiency (SCID)". Frontiers in Pediatrics. 7: 373. doi:10.3389/fped.2019.00373. ISSN 2296-2360. PMC 6759820. PMID 31620409.
- ↑ "National Newborn Screening Status Report" (PDF). Archived from the original (PDF) on 2019-11-16. Retrieved 2022-01-14.
- ↑ "New start date for NHS SCID screening evaluation in England - PHE Screening". phescreening.blog.gov.uk. Archived from the original on 2021-07-16. Retrieved 2021-06-21.
- 1 2 Chinen J, Buckley RH (2010). "Transplantation immunology: solid organ and bone marrow". J. Allergy Clin. Immunol. 125 (2 Suppl 2): S324-35.
- ↑ Vickers, Peter S. (2009). Severe combined immune deficiency: early hospitalisation and isolation. Hoboken NJ: John Wiley & Sons, 29-47. ISBN 978-0-470-74557-1.
- ↑ Buckley RH (2004). "Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution". Annu. Rev. Immunol. 22 (1): 625–655. doi:10.1146/annurev.immunol.22.012703.104614. PMID 15032591.
- 1 2 Fischer A, Hacein-Bey S, Cavazzana-Calvo M (2002). "Gene therapy of severe combined immunodeficiencies". Nat Rev Immunol. 2 (8): 615–621. doi:10.1038/nri859. PMID 12154380. S2CID 39791932.
- ↑ Press release Archived 2007-09-29 at the Wayback Machine from the European Society of Gene Therapy
- ↑ Cavazzana-Calvo M, Fischer A (2007). "Gene therapy for severe combined immunodeficiency: are we there yet?"". J. Clin. Invest. 117 (6): 1456–1465. doi:10.1172/jci30953. PMC 1878528. PMID 17549248.
- ↑ Cavazzana-Calvo, Marina; Fischer, Alain; Hacein-Bey-Abina, Salima; Aiuti, Alessandro (October 2012). "Gene therapy for primary immunodeficiencies: Part 1". Current Opinion in Immunology. 24 (5): 580–584. doi:10.1016/j.coi.2012.08.008. ISSN 1879-0372. PMID 22981681. Archived from the original on 2021-10-08. Retrieved 2022-01-14.
- ↑ "Why Gene Therapy Caused Leukemia In Some 'Boy In The Bubble Syndrome' Patients". ScienceDaily. Archived from the original on 2021-07-19. Retrieved 2021-07-19.
- ↑ Mamcarz, Ewelina; Zhou, Sheng; Lockey, Timothy; Abdelsamed, Hossam; Cross, Shane J.; Kang, Guolian; Ma, Zhijun; Condori, Jose; Dowdy, Jola; Triplett, Brandon; Li, Chen (2019-04-18). "Lentiviral Gene Therapy Combined with Low-Dose Busulfan in Infants with SCID-X1". New England Journal of Medicine. 380 (16): 1525–1534. doi:10.1056/NEJMoa1815408. ISSN 0028-4793. PMC 6636624. PMID 30995372. Archived from the original on 2022-02-07. Retrieved 2022-01-14.
- ↑ "HIV used to cure 'bubble boy' disease". BBC News. 2019-04-17. Archived from the original on 2021-07-19. Retrieved 2021-07-19.
- ↑ Pittman, Jessica Ravitz,John David. "These Scientists May Have Found a Cure for 'Bubble Boy' Disease". Smithsonian Magazine. Archived from the original on 2021-07-19. Retrieved 2021-07-19.
- ↑ Kohn, Donald B.; Booth, Claire; Shaw, Kit L.; Xu-Bayford, Jinhua; Garabedian, Elizabeth; Trevisan, Valentina; Carbonaro-Sarracino, Denise A.; Soni, Kajal; Terrazas, Dayna; Snell, Katie; Ikeda, Alan (2021-05-27). "Autologous Ex Vivo Lentiviral Gene Therapy for Adenosine Deaminase Deficiency". New England Journal of Medicine. 384 (21): 2002–2013. doi:10.1056/NEJMoa2027675. ISSN 0028-4793. PMC 8240285. PMID 33974366. Archived from the original on 2022-02-07. Retrieved 2022-01-14.
- ↑ says, Chris (2021-05-11). "AIDS virus used in gene therapy to fix 'bubble baby' disease". STAT. Archived from the original on 2021-07-19. Retrieved 2021-07-19.
- ↑ "Gene therapy restores immune function in children with rare immunodeficiency". National Institutes of Health (NIH). 2021-05-11. Archived from the original on 2021-07-10. Retrieved 2021-07-19.
- ↑ Tamaroff MH, Nir Y, Straker N (1986). "Children reared in a reverse isolation environment: effects on cognitive and emotional development". J. Autism Dev. Disord. 16 (4): 415–424. doi:10.1007/bf01531708. PMID 3804957. S2CID 30045420.
- ↑ Van der Burg, M; Gennery, AR (2011). "Educational paper. The expanding clinical and immunological spectrum of severe combined immunodeficiency". Eur. J. Pediatr. 170 (5): 561–571. doi:10.1007/s00431-011-1452-3. PMC 3078321. PMID 21479529.
- ↑ "Newborn Screening for Primary Immunodeficiency Disease". Archived from the original on 2016-08-22. Retrieved 2022-01-14.
- ↑ Yee A, De Ravin SS, Elliott E, Ziegler JB (2008). "Severe combined immunodeficiency: A national surveillance study". Pediatr Allergy Immunol. 19 (4): 298–302. doi:10.1111/j.1399-3038.2007.00646.x. PMID 18221464. S2CID 26379956.
- ↑ Yeganeh M, Heidarzade M, Pourpak Z, Parvaneh N, Rezaei N, Gharagozlou M, Movahed M, Shabestari MS, Mamishi S, Aghamohammadi A, Moin M (2008). "Severe combined immunodeficiency: A cohort of 40 patients". Pediatr Allergy Immunol. 19 (4): 303–306. doi:10.1111/j.1399-3038.2007.00647.x. PMID 18093084. S2CID 29466366.
- ↑ El-Maataoui O, Ailal F, Naamane H, Benhsaien I, Jeddane L, Farouqi B, Benslimane A, Jilali N, Oudghiri M, Bousfiha A (2011). "Immunophenotyping of severe combined immunodeficiency in Morocco". IBS J Sci. 26 (4): 161–164. doi:10.1016/j.immbio.2011.05.002.
- ↑ "FOAL.org, an organization promoting research into genetic lethal diseases in horse". Archived from the original on 2008-05-29. Retrieved 2022-03-14.
- ↑ ""The New DNA Test for Severe Combined Immunodeficiency (SCID) in Arabian Horses"". Archived from the original on 2021-02-27. Retrieved 2022-01-14.
- ↑ Henthorn PS, Somberg RL, Fimiani VM, Puck JM, Patterson DF, Felsburg PJ (1994). "IL-2R gamma gene microdeletion demonstrates that canine X-linked severe combined immunodeficiency is a homologue of the human disease". Genomics. 23 (1): 69–74. doi:10.1006/geno.1994.1460. PMID 7829104.
- ↑ Perryman LE (2004). "Molecular pathology of severe combined immunodeficiency in mice, horses, and dogs". Vet. Pathol. 41 (2): 95–100. doi:10.1354/vp.41-2-95. PMID 15017021. S2CID 38273912.
- ↑ Owen, Judith; Punt, Jenni (2013). Kuby Immunology. New York: W.H. Freeman and Company.
- ↑ ""CONVERSION OF NORMAL RATS INTO SCID-LIKE ANIMALS BY MEANS OF BONE MARROW TRANSPLANTATION FROM SCID DONORS ALLOWS ENGRAFTMENT OF HUMAN PERIPHERAL BLOOD MONONUCLEAR CELLS"". Archived from the original on 2021-05-12. Retrieved 2022-01-14.
Further reading
- Buckley RH (2004). "Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution". Annu Rev Immunol. 22: 625–55. doi:10.1146/annurev.immunol.22.012703.104614. PMID 15032591.
- Chinen J, Puck JM (2004). "Successes and risks of gene therapy in primary immunodeficiencies". J Allergy Clin Immunol. 113 (4): 595–603, quiz 604. doi:10.1016/j.jaci.2004.01.765. PMID 15100660. Archived from the original on 2021-10-08. Retrieved 2022-01-14.
- Church AC (2002). "X-linked severe combined immunodeficiency". Hosp Med. 63 (11): 676–80. doi:10.12968/hosp.2002.63.11.1914. PMID 12474613.
- Gennery AR, Cant AJ (2001). "Diagnosis of severe combined immunodeficiency". J Clin Pathol. 54 (3): 191–5. doi:10.1136/jcp.54.3.191. PMC 1731376. PMID 11253129.
External links
- Learning About Severe Combined Immunodeficiency (SCID) Archived 2019-04-02 at the Wayback Machine NIH
| Classification | |
|---|---|
| External resources |