X-linked severe combined immunodeficiency
| X-linked severe combined immunodeficiency | |
|---|---|
| Other names: X-SCID | |
.svg.png.webp) | |
| X-linked recessive is the inheritance pattern of this disorder | |
X-linked severe combined immunodeficiency (X-SCID) is an immunodeficiency disorder in which the body produces very few T cells and NK cells.
In the absence of T cell help, B cells become defective.[1] It is an X-linked recessive inheritance trait, stemming from a mutated (abnormal) version of the IL2RG gene located on the X-chromosome. This gene encodes the interleukin receptor common gamma chain protein, which is a cytokine receptor sub-unit that is part of the receptors for IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21.[2][3]
Symptoms and signs
People with X-SCID often have infections very early in life, before three months of age. This occurs due to the decreased amount of immunoglobulin G (IgG) levels in the infant during the three-month stage.[4] This is followed by viral infections such as pneumonitis, an inflammation of the lung which produces common symptoms such as cough, fever, chills, and shortness of breath.[5] A telltale sign of X-SCID is candidiasis, a type of fungal infection caused by Candida albicans.[6] Candidiasis involves moist areas of the body such as skin, the mouth, respiratory tract, and vagina; symptoms of oral candidiasis include difficulty in swallowing, pain on swallowing and oral lesions. Recurrent eczema-like rashes are also a common symptom. Other common infections experienced by individuals with X-SCID include diarrhea, sepsis, and otitis media.[4] Some other common symptoms that are experienced by X-SCID patients include failure to thrive, gut problems, skin problems, and muscle hypotonia.[4]
In some patients symptoms may not appear for the first six months after birth.[6] This is likely due to passive immunity received from the mother in order to protect the baby from infections until the newborn is able to make its own antibodies.[6] As a result, there can be a silent period where the baby displays no symptoms of X-SCID followed by the development of frequent infections.
Genetics
X-SCID is caused by a mutation occurring in the xq13.1 locus of the X-chromosome.[7] Most often, this disease affects males whose mother is a carrier (heterozygous) for the disorder. Because females have two X-chromosomes, the mother will not be affected by carrying only one abnormal X-chromosome, but any male children will have a 50% chance of being affected with the disorder by inheriting the faulty gene. Likewise, her female children will have a 50% chance of being carriers for the immunodeficiency. X-SCID can also arise through de novo mutations and can be prevented in females by X-inactivation. In X-inactivation the preferential selection of the non-mutant X chromosome during development results in the outcome that none of the mature female cells actively express the X-SCID mutation, they are immunologically unaffected and have no carrier burden. A de novo mutation is an alteration in a gene caused by the result of a mutation in a germ cell (egg or sperm) or in the fertilized egg itself, rather than having been inherited from a carrier. Since only 1/3 of all X-SCID patients have a positive family history of SCID, it is hypothesized that de novo mutations account for a significant percentage of cases.[8] X-inactivation occurs in a completely random manner, in females, very early in embryonic development. Once an X is inactivated, it remains inactivated throughout the life of that cell and any of its daughter cells. It is important to note that X-inactivation is reversed in female germline cells, so that all new oocytes receive an active X. Regardless of which X is inactivated in her somatic cells, a female will have a 50% chance of passing on the disease to any male children.[9]
Pathophysiology
Interleukins are produced by lymphocytes, among other cell types, and are released in response to antigenic and non-antigenic stimuli. The gene IL2RG codes for the common gamma chain protein, which is a common subunit of the individual receptors for Interleukin 2, Interleukin 4, Interleukin 7, Interleukin 9, Interleukin 15 and Interleukin 21.[10] Signalling from these receptors normally promotes growth and differentiation of T-cells, B cells, natural killer cells, glial cells, and cells of the monocyte lineage, depending on the cell type and receptor activated.[11] The most important receptors for X-SCID are those for Interleukin 2, Interleukin 4, Interleukin 7, and Interleukin 15. Specifically, Interleukin 2 and Interleukin 7 are responsible for T-cell proliferation and survival.[12] Likewise, the action of Interleukin 4 and Interleukin 15 will lead to proliferation and differentiation of B-cells into antibody secreting plasma cells.[12] Lastly, Interleukin 15 helps generate developed and matured natural killer cells.[5]
The gene that encodes the common gamma chain in these interleukin receptors is mutated in X-SCID. The mutation leads to an absent or abnormally functioning common gamma chain. The mutation can occur through large, or even single nucleotide, deletions in the IL2RG gene, that disable the common gamma chain so that it is unable to bind with other receptor subunits and signal cytokine activation.[11] Normally, when the interleukin binds to the trimeric receptor protein containing the alpha, beta, and gamma subunits, the common gamma subunit activates Janus Kinase 3 (JAK3), which leads to the phosphorylation of Signal Transducer and Activator of Transcription 5, STAT5. The STAT5 proteins dimerize and translocate to the nucleus, controlling subsequent downstream signalling.[1] Due to the fact that the common gamma chain is absent or abnormal, this downstream pathway is inhibited. This change prevents the T-lymphocytes from signaling other cells, like B-lymphocytes and natural killer cells. Because these cells never receive these signals, they can never mature and differentiate into full grown immune cells.
Diagnosis
Diagnosis of X-SCID is possible through lymphocyte cell counts, lymphocyte function tests, and genetic testing. A healthy immune system should contain large amounts of lymphocytes, but individuals with X-SCID will contain unusually small amounts of T-cells, non-functional B-cells, and some natural killer cells.[9]
| Cell type[9] | Normal lymphocyte count average (range) | X-SCID count average (range) |
|---|---|---|
| T-cells | 3,680 (2,500–5,500) | 200 (0-800) |
| B-cells | 730 (300–2,000) | 1,300 (44 - >3,000) |
| NK cells | 420 (170–1,100) | <100 |
| Total | 0–3 months: 5,400 (3,400–7,300) | <2,000 |
Individuals with X-SCID often have decreased lymphocyte function. This can be tested through the introduction of agents to the immune system; the reaction of the lymphocytes is then observed. In X-SCID, Antibody responses to introduced vaccines and infections are absent, and T-cell responses to mitogens, substances that stimulate lymphocyte transformation, are deficient. IgA and IgM immunoglobulins, substances that aid in fighting off infections, are very low.
The absence of a thymic shadow on chest X-rays is also indicative of X-SCID.[9] In a normal child, a distinctive sailboat shaped shadow near the heart can be seen.[6] The thymus gland in normal patients will gradually decrease in size because the need for the thymus gland diminishes. The decrease in the size of the thymus gland occurs because the body already has a sufficient number of developed T-cells.[13] However, a patient with X-SCID will be born with an abnormally small thymus gland at birth.[9] This indicates that the function of thymus gland, of forming developed T-cells, has been impaired.
Since the mutation in X-SCID is X-linked, there are genetic tests for detecting carriers in X-SCID pedigrees. One method is to look for family-specific IL2RG mutations. Finally, if none of those options are available, there is an unusual pattern of nonrandom X-chromosome inactivation on lymphocytes in carriers, thus looking for such inactivation would prove useful.
If a mother is pregnant and the family has a known history of immunodeficiency, then doctors may perform diagnostic assessment in-utero. Chorionic Villus Sampling, which involves sampling of the placental tissue using a catheter inserted through the cervix, can be performed 8 to 10 weeks into gestation.[14] Alternatively, Amniocentesis, which entails extracting a sample of the fluid which surrounds the fetus, can be performed 15 to 20 weeks into gestation.[14]
Early detection of X-SCID (and other types of SCID) is also made possible through detection of T-cell recombination excision circles, or TRECs. TRECs are composed of excised DNA fragments which are generated during normal splicing of T-cell surface antigen receptors and T-cell maturation.[15] This maturation process is absent across all SCID variants, as evidenced by the low counts of T-lymphocytes. The assay is performed using dried blood from a Guthrie card, from which DNA is extracted.[16] Quantitative PCR is then performed and the number of TRECs determined.[17] Individuals who have the SCID phenotype will have TREC counts as low as <30, compared to approximately 1020 for a healthy infant.[18] A low TREC count indicates that there is insufficient development of T-cells in the thymus gland.[19] This technique can predict SCID even when lymphocyte counts are within the normal range. Newborn screening of X-SCID based on TREC count in dried blood samples has recently been introduced in several states in the United States including California, Colorado, Connecticut, Delaware, Florida, Massachusetts, Michigan, Minnesota, Mississippi, New York, Texas, and Wisconsin.[20] In addition, pilot trials are being performed in several other states beginning in 2013.[21]
Treatments
Treatment for X-linked SCID can be divided into two main groups, the prophylactic treatment (i.e. preventative) and curative treatment.[23] The former attempts to manage the opportunistic infections common to SCID patients[23] and the latter aims at reconstituting healthy T-lymphocyte function.[24]
From the late 60s to early 70s, physicians began using "bubbles", which were plastic enclosures used to house newborns suspected to have SCIDS, immediately after birth.[25] The bubble, a form of isolation, was a sterile environment which meant the infant would avoid infections caused by common and lethal pathogens.[25] On the other hand, prophylactic treatments used today for X-linked SCID are similar to those used to treat other primary immunodeficiencies.[24] There are three types of prophylactic treatments, namely, the use of medication, sterile environments, and intravenous immunoglobulin therapy (IVIG).[24] First, antibiotics or antivirals are administered to control opportunistic infections, such as fluconazole for candidiasis, and acyclovir to prevent herpes virus infection.[26] In addition, the patient can also undergo intravenous immunoglobulin (IVIG) supplementation.[27] Here, a catheter is inserted into the vein and a fluid, containing antibodies normally made by B-cells, is injected into the patient's body.[28] Antibodies, Y-shaped proteins created by plasma cells, recognize and neutralize any pathogens in the body.[29] However, the IVIG is expensive, in terms of time and finance.[30] Therefore, the aforementioned treatments only prevent the infections, and are by no means a cure for X-linked SCID.[24]
Bone marrow transplantation (BMT) is a standard curative procedure and results in a full immune reconstitution, if the treatment is successful.[31] Firstly, a bone marrow transplant requires a human leukocyte antigen (HLA) match between the donor and the recipient.[32] The HLA is distinct from person to person, which means the immune system utilizes the HLA to distinguish self from foreign cells.[33] Furthermore, a BMT can be allogenic or autologous, which means the donor and recipient of bone marrow can be two different people or the same person, respectively.[32] The autologous BMT involves a full HLA match, whereas, the allogenic BMT involves a full or half (haploidentical) HLA match.[34] Particularly, in the allogenic BMT the chances of graft-versus-host-disease occurring is high if the match of the donor and recipient is not close enough.[33] In this case, the T-cells in the donor bone marrow attack the patient's body because the body is foreign to this graft.[35] The depletion of T-cells in the donor tissue and a close HLA match will reduce the chances of graft-versus-host disease occurring.[36] Moreover, patients who received an exact HLA match had normal functioning T-cells in fourteen days.[37] However, those who received a haploidentical HLA match, their T-cells started to function after four months.[37] In addition, the reason BMT is a permanent solution is because the bone marrow contains multipotent hematopoietic stem cells[31] which become common lymphoid or common myeloid progenitors.[38] In particular, the common lymphoid progenitor gives rise to the lymphocytes involved in the immune response (B-cell, T-cell, natural killer cell).[38] Therefore, a BMT will result in a full immune reconstitution but there are aspects of BMT that need to be improved (i.e. GvHD).[39]
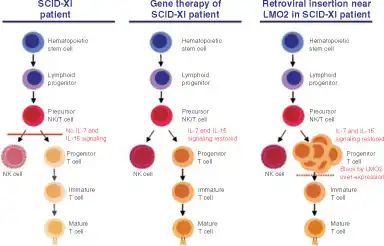
Gene therapy is another treatment option which is available only for clinical trials.[35] X-linked SCID is a monogenic disorder, the IL2RG gene is mutated, so gene therapy will replace this mutated gene with a normal one.[40] This will result in a normal functioning gamma chain protein of the interleukin receptor.[36] In order to transfer a functional gene into the target cell, viral or non-viral vectors can be employed.[36] Viral vectors, such as the retrovirus, that incorporate the gene into the genome result in long-term effects.[35] This, coupled with the bone marrow stem cells, has been successful in treating individuals with X-SCID.[41] In one particular trial by Cavazzana-Calvo et al., ten children were treated with gene therapy at infancy for X-SCID.[42] Nine of the ten were cured of X-SCID.[42] However, about three years after treatment, two of the children developed T-cell leukemia due to insertion of the IL2RG gene near the LMO2 gene and thereby activating the LMO2 gene (a known oncogene).[43] A third child developed leukemia within two years of that study being published, likely as a direct result of the therapy.[44] This condition is known as insertional mutagenesis, where the random insertion of a gene interferes with the tumor suppressor gene or stimulates an oncogene.[35] There is currently no approved gene therapy on the market, but there are many clinical trials into which X-SCID patients may enroll. Therefore, research in the field of gene therapy today and in the future is needed to avoid the occurrence of leukemia.[36] In particular, research into the use of insulator and suicide genes is warranted as this may prevent cancer from developing.[35] The insulator gene inhibits the activation of adjacent genes. On the other hand, the suicide gene is stimulated when a tumour begins to form, and this will result in the deactivation of the therapeutic gene.[35] Moreover, the use of restriction enzymes such as the zinc-finger nuclease (ZFN) is being studied.[35] The ZFN allows the researcher to choose the site of gene integration.[35] Vector safety is important in the field of gene therapy, hence vectors that self-inactivate the promoter and enhancer (SIN) and adenoviruses that creates no immune response are prominent areas of research for vector biologists.[35]
Prognosis
X-linked SCID is a known pediatric emergency which primarily affects males.[32] If the appropriate treatment such as intravenous immunoglobulin supplements, medications for treating infections or a bone marrow transplant is not administered, then the prognosis is poor.[17] The patients with X-linked SCID usually die two years after they are born.[39] For this reason, the diagnosis of X-linked SCID needs to be done early to prevent any pathogens from infecting the infant.
However, the patients have a higher chance of survival if the diagnosis of X-linked SCID is done as soon as the baby is born.[17] This involves taking preventative measures to avoid any infections that can cause death. For example, David Vetter had a high chance of having X-linked SCID because his elder sibling had died due to SCID.[45] This allowed the doctors to place David in the bubble and prevented infections.[45] In addition, if X-linked SCID is known to affect a child, then live vaccines should not be administered and this can save the infant's life. Live attenuated vaccines, which consist of weakened pathogens inserted into the body to create an immune response, can lead to death in infants with X-linked SCID.[46] Moreover, with proper treatments, such as a bone marrow transplant, the prognosis is good. The bone marrow transplant has been successful in treating several patients and resulted in a full immune reconstitution and the patient can live a healthy life.[47] The results of bone marrow transplant are most successful when the closest human leukocyte antigen match has been found.[48] If a close match is not found, however, there is a chance of graft-versus-host-disease which means the donor bone marrow attacks the patient's body.[36] Hence, a close match is required to prevent any complications.
Epidemiology
There is no information on birth ratios/rates, but "X-Linked SCID is the most common form of SCID and it has been estimated to account for 46% to 70% of all SCID cases."[49]
See also
Notes and references
- 1 2 Fisher, A.; Hacein-Bey, S.; Cavazzana-Calvo, M. (August 2002). "Gene therapy of severe combined immunodeficiencies". Nature Reviews Immunology. 2 (8): 615–621. doi:10.1038/nri859. PMID 12154380. S2CID 39791932.
- ↑ Buckley, R.H. (2000). "Advances in the Understanding and Treatment of Human Severe Combined Immunodeficiency". Immunologic Research. 22 (2–3): 237–251. doi:10.1385/ir:22:2-3:237. PMID 11339359. S2CID 36855063.
- ↑ Puck, J.M.; de Saint Basil, G.; Schwarz, K.; Fugmann, S.; Fischer, R.E. (November 1996). "IL2RGbase: a database of γc-chain defects causing human X-SCID". Immunology Today. 17 (11): 507–511. doi:10.1016/0167-5699(96)30062-5. PMID 8961626.
- 1 2 3 Vickers, Peter (2009). Severe Combined Immune Deficiency: Early Hospitalisation and Isolation. Wiley. pp. 29–47. ISBN 978-0-470-31986-4.
- 1 2 Sponzilli, Ivonne; Notarangelo, Luigi D. (2011). "Severe Combined Immunodeficiency (SCID): from molecular basis to clinical management". Acta Bio-medica: Atenei Parmensis. 82 (1): 5–13. PMID 22069950.
- 1 2 3 4 Gennery, A.R.; Cant, A.J. (2001). "Diagnosis of severe combined immunodeficiency". Journal of Clinical Pathology. 54 (3): 191–5. doi:10.1136/jcp.54.3.191. PMC 1731376. PMID 11253129.
- ↑ Buckley, Rebecca H. (1 April 2004). "Molecular Defects in Human Severe Combined Immunodeficiency and Approaches to Immune Reconstitution". Annual Review of Immunology. 22 (1): 625–655. doi:10.1146/annurev.immunol.22.012703.104614. PMID 15032591.
- ↑ Shwartz, R.A. "Pediatric Severe Combined Immunodeficiency". MedScape. Archived from the original on May 24, 2022. Retrieved January 18, 2012.
- 1 2 3 4 5 GeneReviews 2016
- ↑ Cavazzana-Calvo, Marina; Fischer, A. (June 2007). "Gene therapy for severe combined immunodeficiency: are we there yet?". The Journal of Clinical Investigation. 117 (6): 1456–65. doi:10.1172/jci30953. PMC 1878528. PMID 17549248.
- 1 2 Spolski, Rosanne; Leonard, Warren J. (1 April 2008). "Interleukin-21: Basic Biology and Implications for Cancer and Autoimmunity*". Annual Review of Immunology. 26 (1): 57–79. doi:10.1146/annurev.immunol.26.021607.090316. PMID 17953510. Archived from the original on 9 June 2022. Retrieved 29 January 2023.
- 1 2 Leonard, Warren J. (December 2001). "Cytokines and immunodeficiency diseases". Nature Reviews Immunology. 1 (3): 200–8. doi:10.1038/35105066. PMID 11905829. S2CID 5466985. Archived from the original on 2022-03-31. Retrieved 2023-01-29.
- ↑ Rehan, Kelly M. "An Overview of the Thymus: The Gland that Protects You Long after It's Gone". EndocrineWeb. Archived from the original on 2023-04-29. Retrieved 2023-01-29.
- 1 2 Fischer, A. (2000). "Severe combined immunodeficiencies". Clinical & Experimental Immunology. 122 (2): 143–9. doi:10.1046/j.1365-2249.2000.01359.x. PMC 1905779. PMID 11091267.
- ↑ Puck, Jennifer M. (1 March 2012). "Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: The winner is T-cell receptor excision circles". Journal of Allergy and Clinical Immunology. 129 (3): 607–616. doi:10.1016/j.jaci.2012.01.032. PMC 3294074. PMID 22285280.
- ↑ Shearer, William T. (1 March 2012). "Screening for severe combined immunodeficiency in newborns". Journal of Allergy and Clinical Immunology. 129 (3): 619–621. doi:10.1016/j.jaci.2011.12.992. PMID 22277197.
- 1 2 3 Verbsky, James W.; Baker, Mei W.; Grossman, William J.; Hintermeyer, Mary; Dasu, Trivikram; Bonacci, Benedetta; Reddy, Sreelatha; Margolis, David; Casper, James; Gries, Miranda; DeSantes, Ken; Hoffman, Gary L.; Brokopp, Charles D.; Seroogy, Christine M.; Routes, John M. (10 November 2011). "Newborn Screening for Severe Combined Immunodeficiency; The Wisconsin Experience (2008–2011)". Journal of Clinical Immunology. 32 (1): 82–88. doi:10.1007/s10875-011-9609-4. PMID 22068910. S2CID 14226544.
- ↑ Chan, K.; Puck, J. M. (2005). "Development of population-based newborn screening for severe combined immunodeficiency". Journal of Allergy and Clinical Immunology. 115 (2): 391–8. doi:10.1016/j.jaci.2004.10.012. PMID 15696101. Archived from the original on 2023-07-27. Retrieved 2023-01-29.
- ↑ Puck, Jennifer M.; Routes, Jack; Filipovich, Alexandra H.; Sullivan, Kate (20 October 2011). "Expert Commentary: Practical Issues in Newborn Screening for Severe Combined Immune Deficiency (SCID)". Journal of Clinical Immunology. 32 (1): 36–38. doi:10.1007/s10875-011-9598-3. PMC 4380147. PMID 22012274.
- ↑ "IDF SCID Newborn Screening Campaign". Immune Deficiency Foundation. Archived from the original on 2013-05-01. Retrieved 2023-01-29.
- ↑ Wilkerson, Sarah. "Updates from Washington – Day 2". Save Babies Through Screening Foundation. Archived from the original on 2017-06-09. Retrieved 2023-01-29.
- ↑ Pike-Overzet, Karin; van der Burg, Mirjam; Wagemaker, Gerard; van Dongen, Jacques J. M.; Staal, Frank J. T. (November 2007). "New insights and unresolved issues regarding insertional mutagenesis in X-linked SCID gene therapy". Molecular Therapy: The Journal of the American Society of Gene Therapy. 15 (11): 1910–1916. doi:10.1038/sj.mt.6300297. ISSN 1525-0016.
- 1 2 Fischer, A. (2000). "Severe Combined Immunodeficiencies (SCID)". Clinical & Experimental Immunology. 122 (2): 143–9. doi:10.1046/j.1365-2249.2000.01359.x. PMC 1905779. PMID 11091267.
- 1 2 3 4 Rans, T.S.; England, R. (2009). "The evolution of gene therapy in X-linked severe combined immunodeficiency". Annals of Allergy, Asthma & Immunology. 102 (5): 357–363. doi:10.1016/S1081-1206(10)60504-2. PMID 19492655.
- 1 2 Johnston Jr., R.B. (2006). "The Boy in the Bubble". JAMA: The Journal of the American Medical Association. 296 (4): 453–4. doi:10.1001/jama.296.4.453.
- ↑ Freeman, A. F.; Holland, S. M. (2009). "Antimicrobial prophylaxis for primary immunodeficiencies". Current Opinion in Allergy and Clinical Immunology. 9 (6): 525–530. doi:10.1097/ACI.0b013e328332be33. PMID 19812481. S2CID 205434961. Archived from the original on 2023-07-27. Retrieved 2023-01-29.
- ↑ Nolte, M. T.; Pirofsky, B.; Gerritz, G. A.; Golding, B. (1979). "Intravenous immunoglobulin therapy for antibody deficiency". Clinical and Experimental Immunology. 36 (2): 237–43. PMC 1537711. PMID 477026.
- ↑ Farrington, E.; Hochwald, C. (1996). "Intravenous immunoglobulin". Pediatric Nursing. 22 (4): 344–7. PMID 8852117.
- ↑ Shishido, S.N.; Varahan, S.; Yuan, K.; Li, X.; Fleming, S. D. (2012). "Humoral innate immune response and disease". Clinical Immunology. 144 (2): 142–158. doi:10.1016/j.clim.2012.06.002. hdl:2097/16670. PMC 3576926. PMID 22771788.
- ↑ "I.V. immunoglobulin therapy for infectious diseases". Drug and Therapeutics Bulletin. 48 (5): 57–60. 6 May 2010. doi:10.1136/dtb.2009.07.0032. PMID 20447982. S2CID 26258054.
- 1 2 Burg, Mirjam; Gennery, Andy R. (9 April 2011). "Educational paper: The expanding clinical and immunological spectrum of severe combined immunodeficiency". European Journal of Pediatrics. 170 (5): 561–571. doi:10.1007/s00431-011-1452-3. PMC 3078321. PMID 21479529.
- 1 2 3 Buckley, Rebecca H. (15 November 2004). "The multiple causes of human SCID". Journal of Clinical Investigation. 114 (10): 1409–11. doi:10.1172/JCI23571. PMC 525750. PMID 15545990.
- 1 2 Martelli, Massimo F; Aversa, Franco; Bachar-Lustig, Ester; Velardi, Andrea; Reich-Zelicher, Shlomit; Tabilio, Antonio; Gur, Hilit; Reisner, Yair (1 January 2002). "Transplants across human leukocyte antigen barriers". Seminars in Hematology. 39 (1): 48–56. doi:10.1053/shem.2002.29255. PMID 11799529.
- ↑ de la Morena MT, Wayne AS, Day NK, Haag MM, Hinds-Frey KR, Nelson RP, Sutcliffe MJ, Good RA (December 1995). "Recipient T-cell immune reconstitution in X-linked SCID after haploidentical maternal bone marrow transplant". Ann. N. Y. Acad. Sci. 770 (1): 376–7. Bibcode:1995NYASA.770..376M. doi:10.1111/j.1749-6632.1995.tb31074.x. PMID 8597380. S2CID 35195857.
- 1 2 3 4 5 6 7 8 9 Kohn, Donald B.; Sadelain, Michel; Glorioso, Joseph C. (1 July 2003). "Occurrence of leukaemia following gene therapy of X-linked SCID". Nature Reviews Cancer. 3 (7): 477–488. doi:10.1038/nrc1122. PMID 12835668. S2CID 62790581.
- 1 2 3 4 5 Rans, Tonya S.; England, Ronald (1 May 2009). "The evolution of gene therapy in X-linked severe combined immunodeficiency". Annals of Allergy, Asthma & Immunology. 102 (5): 357–363. doi:10.1016/S1081-1206(10)60504-2. PMID 19492655.
- 1 2 Buckley, Rebecca H.; Schiff, Sherrie E.; Schiff, Richard I.; Markert, M. Louise; Williams, Larry W.; Roberts, Joseph L.; Myers, Laurie A.; Ward, Frances E. (18 February 1999). "Hematopoietic Stem-Cell Transplantation for the Treatment of Severe Combined Immunodeficiency". New England Journal of Medicine. 340 (7): 508–516. doi:10.1056/NEJM199902183400703. PMID 10021471.
- 1 2 Kondo, Motonari; Wagers, Amy J.; Manz, Markus G.; Prohaska, Susan S.; Scherer, David C.; Beilhack, Georg F.; Shizuru, Judith A.; Weissman, Irving L. (1 April 2003). "Biology of Hemaptopoietic Stem Cells and Progenitors: Implications for Clinical Application". Annual Review of Immunology. 21 (1): 759–806. doi:10.1146/annurev.immunol.21.120601.141007. PMID 12615892.
- 1 2 Fischer, A. (1 November 2000). "Severe combined immunodeficiencies (SCID)". Clinical and Experimental Immunology. 122 (2): 143–9. doi:10.1046/j.1365-2249.2000.01359.x. PMC 1905779. PMID 11091267.
- ↑ Buckley, Rebecca H. (15 November 2004). "The multiple causes of human SCID". Journal of Clinical Investigation. 114 (10): 1409–11. doi:10.1172/JCI23571. PMC 525750. PMID 15545990.
- ↑ Woods N-B, Bottero V, Schmidt M, von Kalle C, Verma IM (21 September 2006). "Gene therapy: Is IL2RG oncogenic in T-cell development?: X-SCID transgene leukaemogenicity (reply)". Nature. 443 (7109): E6–E7. Bibcode:2006Natur.443E...6W. doi:10.1038/nature05220. PMID 16988660. S2CID 4418027.
- 1 2 Cavazzana-Calvo, M. (28 April 2000). "Gene Therapy of Human Severe Combined Immunodeficiency (SCID)-X1 Disease". Science. 288 (5466): 669–672. Bibcode:2000Sci...288..669C. doi:10.1126/science.288.5466.669. PMID 10784449.
- ↑ "Gene Therapy Insertional Mutagenesis Insights" (PDF). Science. 16 January 2004. Archived (PDF) from the original on 7 January 2009. Retrieved 29 January 2023.
- ↑ "X-SCID Gene Therapy Poses Substantial Cancer Risk". Medical News Today. 30 April 2006. Archived from the original on 3 January 2009. Retrieved 29 January 2023.
- 1 2 Cossu, Fausto (1 January 2010). "Genetics of SCID". Italian Journal of Pediatrics. 36 (1): 76. doi:10.1186/1824-7288-36-76. PMC 2999594. PMID 21078154.
- ↑ Nima Rezaei; Asghar Aghamohammadi; Luigi Notarangelo, eds. (2008). Primary immunodeficiency diseases : definition, diagnosis, and management. Berlin: Springer. p. 45. ISBN 978-3-540-78537-8.
- ↑ Dvorak, C C; Cowan, M J (29 October 2007). "Hematopoietic stem cell transplantation for primary immunodeficiency disease". Bone Marrow Transplantation. 41 (2): 119–126. doi:10.1038/sj.bmt.1705890. PMID 17968328.
- ↑ Mikkers, Harald; Pike-Overzet, Karin; Staal, Frank J.T. (8 February 2012). "Induced pluripotent stem cells and severe combined immunodeficiency: merely disease modeling or potentially a novel cure?". Pediatric Research. 71 (4–2): 427–432. doi:10.1038/pr.2011.65. PMID 22430378.
- ↑ Davis J, Puck JM (1993). "X-Linked Severe Combined Immunodeficiency". In Pagon RA, Bird TD, Dolan CR, Stephens K (eds.). GeneReviews. Seattle WA: University of Washington. PMID 20301584. Archived from the original on 2023-01-31. Retrieved 2023-01-29.
References
- https://web.archive.org/web/20081122091233/http://www.scid.net/about.htm
- Allenspach E, Rawlings DJ, Scharenberg AM (2016) [1993]. "X-Linked Severe Combined Immunodeficiency". In Pagon RA, Bird TD, Dolan CR, Stephens K (eds.). GeneReviews [Internet]. Seattle WA: University of Washington. PMID 20301584. NBK1410. Archived from the original on 2023-01-31. Retrieved 2023-01-29.
- Online Mendelian Inheritance in Man (OMIM): Interleukin 2 receptor, Gamma; IL2RG - 308380
- Online Mendelian Inheritance in Man (OMIM): Severe Combined Immunodeficiency, X-Linked; SCIDX1 - 300400
External links
| Classification |
|---|