Ataxia–telangiectasia
| Ataxia–telangiectasia | |
|---|---|
| Other names: Louis–Bar syndrome | |
 | |
| Autosomal recessive | |
Ataxia–telangiectasia (AT or A–T), also referred to as ataxia–telangiectasia syndrome or Louis–Bar syndrome,[1][2] is a rare, neurodegenerative, autosomal recessive disease causing severe disability. Ataxia refers to poor coordination and telangiectasia to small dilated blood vessels, both of which are hallmarks of the disease.[3] A–T affects many parts of the body:
- It impairs certain areas of the brain including the cerebellum, causing difficulty with movement and coordination.
- It weakens the immune system, causing a predisposition to infection.
- It prevents repair of broken DNA, increasing the risk of cancer.
Symptoms most often first appear in early childhood (the toddler stage) when children begin to sit or walk. Though they usually start walking at a normal age, they wobble or sway when walking, standing still or sitting. In late pre-school and early school age, they develop difficulty moving their eyes in a natural manner from one place to the next (oculomotor apraxia). They develop slurred or distorted speech, and swallowing problems. Some have an increased number of respiratory tract infections (ear infections, sinusitis, bronchitis, and pneumonia). Because not all children develop in the same manner or at the same rate, it may be some years before A–T is properly diagnosed. Most children with A–T have stable neurologic symptoms for the first 4–5 years of life, but begin to show increasing problems in early school years.
A–T is caused by a defect in the ATM gene,[4] which is involved in the recognition and repair of damaged DNA.[5] The prevalence of A–T is estimated to be as high as 1 in 40,000 to as low as 1 in 300,000 people.[6][7]
Symptoms and signs
There is substantial variability in the severity of features of A–T among affected individuals, and at different ages. The following symptoms or problems are either common or important features of A–T:
- Ataxia (difficulty with control of movement) that is apparent early but worsens in school to pre-teen years
- Oculomotor apraxia (difficulty with coordination of head and eye movement when shifting gaze from one place to the next)
- Involuntary movements
- Telangiectasia (dilated blood vessels) over the white (sclera) of the eyes, making them appear bloodshot. These are not apparent in infancy and may first appear at age 5–8 years. Telangiectasia may also appear on sun-exposed areas of skin.
- Problems with infections, especially of the ears, sinuses and lungs
- Increased incidence of cancer (primarily, but not exclusively, lymphomas and leukemias)
- Delayed onset or incomplete pubertal development, and very early menopause
- Slowed rate of growth (weight and/or height)
- Drooling particularly in young children when they are tired or concentrating on activities
- Dysarthria (slurred, slow, or distorted speech sounds)
- Diabetes in adolescence or later
- Premature changes in hair and skin
Many children are initially misdiagnosed as having cerebral palsy. The diagnosis of A–T may not be made until the preschool years when the neurologic symptoms of impaired gait, hand coordination, speech and eye movement appear or worsen, and the telangiectasia first appear. Because A–T is so rare, doctors may not be familiar with the symptoms, or methods of making a diagnosis. The late appearance of telangiectasia may be a barrier to the diagnosis.[8] It may also take some time before doctors consider A–T as a possibility because of the early stability of symptoms and signs. There are patients who have been diagnosed with A-T only in adulthood due to an attenuated form of the disease, and this has been correlated with the type of their gene mutation.[9][10][11][12]
Ataxia and other neurologic problems
The first indications of A–T usually occur during the toddler years. Children start walking at a normal age, but may not improve much from their initial wobbly gait. Sometimes they have problems standing or sitting still and tend to sway backward or from side to side. In primary school years, walking becomes more difficult, and children will use doorways and walls for support. Children with A–T often appear better when running or walking quickly in comparison to when they are walking slowly or standing in one place. Around the beginning of their second decade, children with the more severe ("classic") form of A–T start using a wheelchair for long distances. During school years, children may have increasing difficulty with reading because of impaired coordination of eye movement. At the same time, other problems with fine-motor functions (writing, coloring, and using utensils to eat), and with slurring of speech (dysarthria) may arise. Most of these neurologic problems stop progressing after the age of about 12 – 15 years, though involuntary movements may start at any age and may worsen over time. These extra movements can take many forms, including small jerks of the hands and feet that look like fidgeting (chorea), slower twisting movements of the upper body (athetosis), adoption of stiff and twisted postures (dystonia), occasional uncontrolled jerks (myoclonic jerks), and various rhythmic and non-rhythmic movements with attempts at coordinated action (tremors).[13][14]
Telangiectasia

Prominent blood vessels (telangiectasia) over the white (sclera) of the eyes usually occur by the age of 5–8 years, but sometimes appear later or not at all.[8] The absence of telangiectasia does not exclude the diagnosis of A–T. Potentially a cosmetic problem, the ocular telangiectasia do not bleed or itch, though they are sometimes misdiagnosed as chronic conjunctivitis. It is their constant nature, not changing with time, weather or emotion, that marks them as different from other visible blood vessels. Telangiectasia can also appear on sun-exposed areas of skin, especially the face and ears. They occur in the bladder as a late complication of chemotherapy with cyclophosphamide,[15] have been seen deep inside the brain of older people with A–T,[16] and occasionally arise in the liver and lungs.[17]
Immune problems
About two-thirds of people with A–T have abnormalities of the immune system.[18] The most common abnormalities are low levels of one or more classes of immunoglobulins (IgG, IgA, IgM, and IgG subclasses), not making antibodies in response to vaccines or infections, and having low numbers of lymphocytes (especially T-lymphocytes) in the blood. Some people have frequent infections of the upper (colds, sinus and ear infections) and lower (bronchitis and pneumonia) respiratory tract. All children with A–T should have their immune systems evaluated to detect those with severe problems that require treatment to minimize the number or severity of infections. Some people with A–T need additional immunizations (especially with pneumonia and influenza vaccines), antibiotics to provide protection (prophylaxis) from infections, and/or infusions of immunoglobulins (gamma globulin). The need for these treatments should be determined by an expert in the field of immunodeficiency or infectious diseases.[17]
Cancer
People with A–T have a highly increased incidence (approximately 25% lifetime risk) of cancers, particularly lymphomas and leukemia, but other cancers can occur.[19]
Women who are A–T carriers (who have one mutated copy of the ATM gene), have approximately a two-fold increased risk for the development of breast cancer compared to the general population.[20][21] This includes all mothers of A–T children and some female relatives. Current consensus is that special screening tests are not helpful, but all women should have routine cancer surveillance.
Skin
A–T can cause features of early aging such as premature graying of the hair. It can also cause vitiligo (an auto-immune disease causing loss of skin pigment resulting in a blotchy “bleach-splashed” look), and warts which can be extensive and recalcitrant to treatment. A small number of people develop a chronic inflammatory skin disease (granulomas).[22]
Lung disease
Chronic lung disease develops in more than 25% of people with A–T.[23]
Lung function tests (spirometry) should be performed at least annually in children old enough to perform them, influenza and pneumococcal vaccines given to eligible individuals, and sinopulmonary infections treated aggressively to limit the development of chronic lung disease.
Feeding, swallowing, and nutrition
Feeding and swallowing can become difficult for people with A–T as they get older.[24]
Involuntary movements may make feeding difficult or messy and may excessively prolong mealtimes. It may be easier to finger feed than use utensils (e.g., spoon or fork). For liquids, it is often easier to drink from a closed container with a straw than from an open cup. Caregivers may need to provide foods or liquids so that self-feeding is possible, or they may need to feed the person with A–T. In general, meals should be completed within approximately 30 minutes. Longer meals may be stressful, interfere with other daily activities, and limit the intake of necessary liquids and nutrients.
If swallowing problems (dysphagia) occur, they typically present during the second decade of life. Dysphagia is common because of the neurological changes that interfere with coordination of mouth and pharynx (throat) movements that are needed for safe and efficient swallowing. Coordination problems involving the mouth may make chewing difficult and increase the duration of meals. Problems involving the pharynx may cause liquid, food, and saliva to be inhaled into the airway (aspiration). People with dysphagia may not cough when they aspirate (silent aspiration). Swallowing problems and especially swallowing problems with silent aspiration may cause lung problems due to inability to cough and clear food and liquids from the airway.
- Warning signs of a swallowing problem
- Choking or coughing when eating or drinking
- Poor weight gain (during ages of expected growth) or weight loss at any age
- Excessive drooling
- Mealtimes longer than 40 – 45 minutes, on a regular basis
- Foods or drinks previously enjoyed are now refused or difficult
- Chewing problems
- Increase in the frequency or duration of breathing or respiratory problems
- Increase in lung infections
Eye and vision
- Most people develop telangiectasia (prominent blood vessels) in the membrane that covers the white part (sclera) of the eye.
- Vision (ability to see objects in focus) is normal.[25]
- Control of eye movement is often impaired, affecting visual functions that require fast, accurate eye movements from point to point (e.g. reading).
- Eye misalignments (strabismus) are common, but may be treatable.
- There may be difficulty in coordinating eye position and shaping the lens to see objects up close.
Orthopedics
Many individuals with A–T develop deformities of the feet that compound the difficulty they have with walking due to impaired coordination. Early treatment may slow progression of this deformity. Bracing or surgical correction sometimes improves stability at the ankle sufficient to enable an individual to walk with support, or bear weight during assisted standing transfers from one seat to another. Severe scoliosis is relatively uncommon, but probably does occur more often than in those without A–T. Spinal fusion is only rarely indicated.
Genetics
.png.webp)
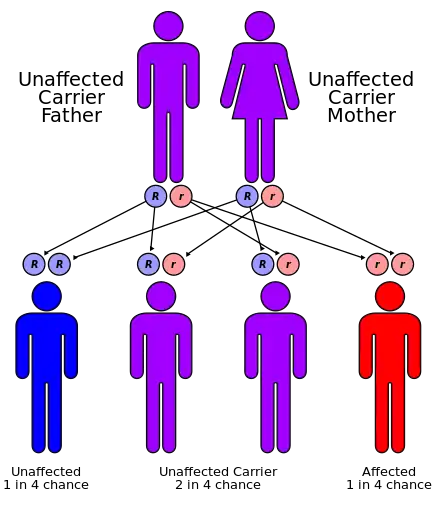
A–T is caused by mutations in the ATM (ATM serine/threonine kinase or ataxia–telangiectasia mutated) gene, which was cloned in 1995.[4] ATM is located on human chromosome 11 (11q22.3) and is made up of 69 exons spread across 150kb of genomic DNA.[26]
The mode of inheritance for A–T is autosomal recessive. Each parent is a carrier, meaning that they have one normal copy of the A–T gene (ATM) and one copy that is mutated. A–T occurs if a child inherits the mutated A–T gene from each parent, so in a family with two carrier parents, there is 1 chance in 4 that a child born to the parents will have the disorder. Prenatal diagnosis (and carrier detection) can be carried out in families if the errors (mutation) in an affected child's two ATM genes have been identified. The process of getting this done can be complicated and, as it requires time, should be arranged before conception.
Looking for mutations in the ATM gene of an unrelated person (for example, the spouse of a known A–T carrier) presents significant challenges. Genes often have variant spellings (polymorphisms) that do not affect function. In a gene as large as ATM, such variant spellings are likely to occur and doctors cannot always predict whether a specific variant will or will not cause disease. Genetic counseling can help family members of an A–T patient understand what can or cannot be tested, and how the test results should be interpreted.
Carriers of A–T, such as the parents of a person with A–T, have one mutated copy of the ATM gene and one normal copy. They are generally healthy, but there is an increased risk of breast cancer in women. This finding has been confirmed in a variety of different ways, and is the subject of current research. Standard surveillance (including monthly breast self-exams and mammography at the usual schedule for age) is recommended, unless additional tests are indicated because the individual has other risk factors (e.g., family history of breast cancer).
Pathophysiology
How does loss of the ATM protein create a multisystem disorder?

A–T has been described as a genome instability syndrome, a DNA repair disorder and a DNA damage response (DDR) syndrome. ATM, the gene responsible for this multi-system disorder, encodes a protein of the same name which coordinates the cellular response to DNA double strand breaks (DSBs).[27] Radiation therapy, chemotherapy that acts like radiation (radiomimetic drugs) and certain biochemical processes and metabolites can cause DSBs. When these breaks occur, ATM stops the cell from making new DNA (cell cycle arrest) and recruits and activates other proteins to repair the damage. Thus, ATM allows the cell to repair its DNA before the completion of cell division. If DNA damage is too severe, ATM will mediate the process of programmed cell death (apoptosis) to eliminate the cell and prevent genomic instability.[28]
Cancer and radiosensitivity
In the absence of the ATM protein, cell-cycle check-point regulation and programmed cell death in response to DSBs are defective. The result is genomic instability which can lead to the development of cancers.[35]
Irradiation and radiomimetic compounds induce DSBs which are unable to be repaired appropriately when ATM is absent. Consequently, such agents can prove especially cytotoxic to A–T cells and people with A–T.
Delayed pubertal development (gonadal dysgenesis)
Infertility is often described as a characteristic of A–T. Whereas this is certainly the case for the mouse model of A–T,[36] in humans it may be more accurate to characterize the reproductive abnormality as gonadal atrophy or dysgenesis characterized by delayed pubertal development. Because programmed DSBs are generated to initiate genetic recombinations involved in the production of sperm and eggs in reproductive organs (a process known as meiosis), meiotic defects and arrest can occur when ATM is not present.[36][37][38]
Immune system defects and immune-related cancers

As lymphocytes develop from stem cells in the bone marrow into mature lymphocytes in the periphery, they rearrange special segments of their DNA [V(D)J recombination process]. This process requires them to make DSBs, which are difficult to repair in the absence of ATM.[43][44][45][46] As a result, most people with A–T have reduced numbers of lymphocytes and some impairment of lymphocyte function (such as an impaired ability to make antibodies in response to vaccines or infections). In addition, broken pieces of DNA in chromosomes involved in the above-mentioned rearrangements have a tendency to recombine with other genes (translocation), making the cells prone to the development of cancer (lymphoma and leukemia).
Progeric changes
Cells from people with A–T demonstrate genomic instability, slow growth and premature senescence in culture, shortened telomeres and an ongoing, low-level stress response.[5][47] These factors may contribute to the progeric (signs of early aging) changes of skin and hair sometimes observed in people with A–T. For example, DNA damage and genomic instability cause melanocyte stem cell (MSC) differentiation which produces graying. Thus, ATM may be a “stemness checkpoint” protecting against MSC differentiation and premature graying of the hair.[48]
Telangiectasia
The cause of telangiectasia or dilated blood vessels in the absence of the ATM protein is not yet known.
Increased alpha-fetoprotein (AFP) levels
Approximately 95% of people with A–T have elevated serum AFP levels after the age of two, and measured levels of AFP appear to increase slowly over time.[49] AFP levels are very high in the newborn, and normally descend to adult levels over the first year to 18 months. The reason why individuals with A–T have elevated levels of AFP is not yet known.
Neurodegeneration
A–T is one of several DNA repair disorders that result in neurological abnormalities or degeneration. Arguably some of the most devastating symptoms of A–T are a result of progressive cerebellar degeneration, characterized by the loss of Purkinje cells and, to a lesser extent, granule cells (located exclusively in the cerebellum).[13] The cause of this cell loss is not known, though many hypotheses have been proposed based on experiments performed both in cell culture and in the mouse model of A–T. Current hypotheses explaining the neurodegeneration associated with A–T include the following:
- Defective DNA damage response in neurons[29][50] which can lead to
- Defective response to oxidative stress characterized by elevated ROS and altered cellular metabolism[32][56][57][58]
- Mitochondrial dysfunction[31][59][60]
- Defects in neuronal function:
- Altered protein turnover[67]
These hypotheses may not be mutually exclusive and more than one of these mechanisms may underlie neuronal cell death when there is an absence or deficiency of ATM. Further, cerebellar damage and loss of Purkinje and granule cells do not explain all of the neurologic abnormalities seen in people with A–T. The effects of ATM deficiency on the other areas of the brain outside of the cerebellum are being actively investigated.
Radiation exposure
People with A–T have an increased sensitivity to ionizing radiation (X-rays and gamma rays). Therefore, X-ray exposure should be limited to times when it is medically necessary, as exposing an A–T patient to ionizing radiation can damage cells in such a way that the body cannot repair them. The cells can cope normally with other forms of radiation, such as ultraviolet light, so there is no need for special precautions from sunlight exposure.
Diagnosis
The diagnosis of A–T is usually suspected by the combination of neurologic clinical features (ataxia, abnormal control of eye movement, and postural instability) with telangiectasia and sometimes increased infections, and confirmed by specific laboratory abnormalities (elevated alpha-fetoprotein levels, increased chromosomal breakage or cell death of white blood cells after exposure to X-rays, absence of ATM protein in white blood cells, or mutations in each of the person's ATM genes).
A variety of laboratory abnormalities occur in most people with A–T, allowing for a tentative diagnosis to be made in the presence of typical clinical features. Not all abnormalities are seen in all patients. These abnormalities include:
- Elevated and slowly increasing alpha-fetoprotein levels in serum after 2 years of age
- Immunodeficiency with low levels of immunoglobulins (especially IgA, IgM, IgG, and IgG subclasses) and low number of lymphocytes in the blood
- Chromosomal instability (broken pieces of chromosomes)
- Increased sensitivity of cells to x-ray exposure (cells die or develop even more breaks and other damage to chromosomes)[68]
- Cerebellar atrophy on MRI scan
The diagnosis can be confirmed in the laboratory by finding an absence or deficiency of the ATM protein in cultured blood cells,[69][70] an absence or deficiency of ATM function (kinase assay), or mutations in both copies of the cell's ATM gene. These more specialized tests are not always needed, but are particularly helpful if a child's symptoms are atypical.
Differential diagnosis
There are several other disorders with similar symptoms or laboratory features that physicians may consider when diagnosing A–T.[71] The three most common disorders that are sometimes confused with A–T are:
- Cerebral palsy
- Friedreich's ataxia
- Cogan oculomotor apraxia
Each of these can be distinguished from A–T by the neurologic exam and clinical history.
Cerebral palsy (CP) describes a non-progressive disorder of motor function stemming from malformation or early damage to the brain. CP can manifest in many ways, given the different manner in which the brain can be damaged; in common to all forms is the emergence of signs and symptoms of impairment as the child develops. However, milestones that have been accomplished and neurologic functions that have developed do not deteriorate in CP as they often do in children with A–T in the late pre-school years. Most children with ataxia caused by CP do not begin to walk at a normal age, whereas most children with A–T start to walk at a normal age even though they often "wobble" from the start. Pure ataxia is a rare manifestation of early brain damage or malformation, however, and the possibility of an occult genetic disorder of brain should be considered and sought for those in whom ataxia is the chief manifestation of CP. Children with ataxic CP will not manifest the laboratory abnormalities associated with A–T.
Cogan occulomotor apraxia is a rare disorder of development. Affected children have difficulty moving their eyes only to a new visual target, so they will turn their head past the target to “drag” the eyes to the new object of interest, then turn the head back. This tendency becomes evident in late infancy and toddler years, and mostly improves with time. This contrasts to the oculomotor difficulties evident in children with A–T, which are not evident in early childhood but emerge over time. Cogan's oculomotor apraxia is generally an isolated problem, or may be associated with broader developmental delay.
Friedreich ataxia (FA) is the most common genetic cause of ataxia in children. Like A–T, FA is a recessive disease, appearing in families without a history of the disorder. FA is caused by mutation in the frataxin gene, most often an expansion of a naturally occurring repetition of the three nucleotide bases GAA from the usual 5–33 repetitions of this trinucleotide sequence to greater than 65 repeats on each chromosome. Most often the ataxia appears between 10 and 15 years of age, and differs from A–T by the absence of telangiectasia and oculomotor apraxia, a normal alpha fetoprotein, and the frequent presence of scoliosis, absent tendon reflexes, and abnormal features on the EKG. Individuals with FA manifest difficulty standing in one place that is much enhanced by closure of the eyes (Romberg sign) that is not so apparent in those with A–T – even though those with A–T may have greater difficulty standing in one place with their eyes open.
There are other rare disorders that can be confused with A–T, either because of similar clinical features, a similarity of some laboratory features, or both. These include:
- Ataxia–oculomotor apraxia type 1 (AOA1)
- Ataxia–oculomotor apraxia type 2 (AOA2 also known as SCAR1)
- Ataxia–telangiectasia like disorder (ATLD)
- Nijmegen breakage syndrome (NBS)

Ataxia–oculomotor apraxia type 1 (AOA1) is an autosomal recessive disorder similar to A–T in manifesting increasing problems with coordination and oculomotor apraxia, often at a similar age to those having A–T. It is caused by mutation in the gene coding for the protein aprataxin. Affected individuals differ from those with A–T by the early appearance of peripheral neuropathy, early in their course manifest difficulty with initiation of gaze shifts, and the absence of ocular telangiectasia, but laboratory features are of key importance in the differentiation of the two. Individuals with AOA1 have a normal AFP, normal measures of immune function, and after 10–15 years have low serum levels of albumin. Genetic testing of the aprataxin gene can confirm the diagnosis. There is no enhanced risk for cancer.
Ataxia–oculomotor apraxia type 2 (AOA2) is an autosomal recessive disorder also similar to A–T in manifesting increasing problems with coordination and peripheral neuropathy, but oculomotor apraxia is present in only half of affected individuals. Ocular telangiectasia do not develop. Laboratory abnormalities of AOA2 are like A–T, and unlike AOA1, in having an elevated serum AFP level, but like AOA1 and unlike A–T in having normal markers of immune function. Genetic testing of the senataxin gene (SETX) can confirm the diagnosis. There is no enhanced risk for cancer.
Ataxia–telangiectasia like disorder (ATLD) is an extremely rare condition, caused by mutation in the hMre11 gene, that could be considered in the differential diagnosis of A–T. Patients with ATLD are very similar to those with A–T in showing a progressive cerebellar ataxia, hypersensitivity to ionizing radiation and genomic instability. Those rare individuals with ATLD who are well described differ from those with A–T by the absence of telangiectasia, normal immunoglobulin levels, a later onset, and a slower progression of the symptoms. Because of its rarity, it is not yet known whether or not ATLD carries an increased risk to develop cancer. Because those mutations of Mre11 that severely impair the MRE11 protein are incompatible with life, individuals with ATLD all have some partial function of the Mre11 protein, and hence likely all have their own levels of disease severity.
Nijmegen breakage syndrome (NBS) is a rare genetic disorder that has similar chromosomal instability to that seen in people with A–T, but the problems experienced are quite different. Children with NBS have significant microcephaly, a distinct facial appearance, short stature, and moderate cognitive impairment, but do not experience any neurologic deterioration over time. Like those with A–T, children with NBS have enhanced sensitivity to radiation, disposition to lymphoma and leukemia, and some laboratory measures of impaired immune function, but do not have ocular telangiectasia or an elevated level of AFP.
The proteins expressed by the hMre11 (defective in ATLD) and Nbs1 (defective in NBS) genes exist in the cell as a complex, along with a third protein expressed by the hRad50 gene. This complex, known as the MRN complex, plays an important role in DNA damage repair and signaling and is required to recruit ATM to the sites of DNA double strand breaks. Mre11 and Nbs1 are also targets for phosphorylation by the ATM kinase. Thus, the similarity of the three diseases can be explained in part by the fact that the protein products of the three genes mutated in these disorders interact in common pathways in the cell.
Differentiation of these disorders is often possible with clinical features and selected laboratory tests. In cases where the distinction is unclear, clinical laboratories can identify genetic abnormalities of ATM, aprataxin and senataxin, and specialty centers can identify abnormality of the proteins of potentially responsible genes, such as ATM, MRE11, nibrin, TDP1, aprataxin and senataxin as well as other proteins important to ATM function such as ATR, DNA-PK, and RAD50.
Management
Ataxia and other neurologic problems
There is no treatment known to slow or stop the progression of the neurologic problems.
Immune problems
All individuals with A–T should have at least one comprehensive immunologic evaluation that measures the number and type of lymphocytes in the blood (T-lymphocytes and B-lymphocytes), the levels of serum immunoglobulins (IgG, IgA, and IgM) and antibody responses to T-dependent (e.g., tetanus, Hemophilus influenzae b) and T-independent (23-valent pneumococcal polysaccharide) vaccines. For the most part, the pattern of immunodeficiency seen in an A–T patient early in life (by age five) will be the same pattern seen throughout the lifetime of that individual. Therefore, the tests need not be repeated unless that individual develops more problems with infection. Problems with immunity sometimes can be overcome by immunization. Vaccines against common bacterial respiratory pathogens such as Hemophilus influenzae, pneumococci and influenza virus (the “flu”) are commercially available and often help to boost antibody responses, even in individuals with low immunoglobulin levels. If the vaccines do not work and the patient continues to have problems with infections, gamma globulin therapy (IV or subcutaneous infusions of antibodies collected from normal individuals) may be of benefit. A small number of people with A–T develop an abnormality in which one or more types of immunoglobulin are increased far beyond the normal range. In a few cases, the immunoglobulin levels can be increased so much that the blood becomes thick and does not flow properly. Therapy for this problem must be tailored to the specific abnormality found and its severity.
If an individual patient's susceptibility to infection increases, it is important to reassess immune function in case deterioration has occurred and a new therapy is indicated. If infections are occurring in the lung, it is also important to investigate the possibility of dysfunctional swallow with aspiration into the lungs (see above sections under Symptoms: Lung Disease and Symptoms: Feeding, Swallowing and Nutrition.)
Most people with A–T have low lymphocyte counts in the blood. This problem seems to be relatively stable with age, but a rare number of people do have progressively decreasing lymphocyte counts as they get older. In the general population, very low lymphocyte counts are associated with an increased risk for infection. Such individuals develop complications from live viral vaccines (measles, mumps, rubella and chickenpox), chronic or severe viral infections, yeast infections of the skin and vagina, and opportunistic infections (such as pneumocystis pneumonia). Although lymphocyte counts are often as low in people with A–T, they seldom have problems with opportunistic infections. (The one exception to that rule is that problems with chronic or recurrent warts are common.) The number and function of T-lymphocytes should be re-evaluated if a person with A–T is treated with corticosteroid drugs such as prednisone for longer than a few weeks or is treated with chemotherapy for cancer. If lymphocyte counts are low in people taking those types of drugs, the use of prophylactic antibiotics is recommended to prevent opportunistic infections.
If the tests show significant abnormalities of the immune system, a specialist in immunodeficiency or infectious diseases will be able to discuss various treatment options. Absence of immunoglobulin or antibody responses to vaccine can be treated with replacement gamma globulin infusions, or can be managed with prophylactic antibiotics and minimized exposure to infection. If antibody function is normal, all routine childhood immunizations including live viral vaccines (measles, mumps, rubella and varicella) should be given. In addition, several “special” vaccines (that is, licensed but not routine for otherwise healthy children and young adults) should be given to decrease the risk that an A–T patient will develop lung infections. The patient and all household members should receive the influenza (flu) vaccine every fall. People with A–T who are less than two years old should receive three doses of a pneumococcal conjugate vaccine (Prevnar) given at two month intervals. People older than two years who have not previously been immunized with Prevnar should receive two doses of Prevnar. At least 6 months after the last Prevnar has been given and after the child is at least two years old, the 23-valent pneumococcal vaccine should be administered. Immunization with the 23-valent pneumococcal vaccine should be repeated approximately every five years after the first dose.
In people with A–T who have low levels of IgA, further testing should be performed to determine whether the IgA level is low or completely absent. If absent, there is a slightly increased risk of a transfusion reaction. “Medical Alert” bracelets are not necessary, but the family and primary physician should be aware that if there is elective surgery requiring red cell transfusion, the cells should be washed to decrease the risk of an allergic reaction.
People with A–T also have an increased risk of developing autoimmune or chronic inflammatory diseases. This risk is probably a secondary effect of their immunodeficiency and not a direct effect of the lack of ATM protein. The most common examples of such disorders in A–T include immune thrombocytopenia (ITP), several forms of arthritis, and vitiligo.
Lung disease
Recurrent sinus and lung infections can lead to the development of chronic lung disease.[23] Such infections should be treated with appropriate antibiotics to prevent and limit lung injury. Administration of antibiotics should be considered when children and adults have prolonged respiratory symptoms (greater than 7 days), even following what was presumed to have been a viral infection. To help prevent respiratory illnesses from common respiratory pathogens, annual influenza vaccinations should be given and pneumococcal vaccines should be administered when appropriate. Antibiotic treatment should also be considered in children with chronic coughs that are productive of mucous, those who do not respond to aggressive pulmonary clearance techniques and in children with muco-purulent secretions from the sinuses or chest. A wet cough can also be associated with chronic aspiration which should be ruled out through proper diagnostic studies, however, aspiration and respiratory infections are not necessarily exclusive of each other. In children and adults with bronchiectasis, chronic antibiotic therapy should be considered to slow chronic lung disease progression.
Culturing of the sinuses may be needed to direct antibiotic therapy. This can be done by an Ear Nose and Throat (ENT) specialist. In addition, diagnostic bronchoscopy may be necessary in people who have recurrent pneumonias, especially those who do not respond or respond incompletely to a course of antibiotics.
Clearance of bronchial secretions is essential for good pulmonary health and can help limit injury from acute and chronic lung infections. Children and adults with increased bronchial secretions can benefit from routine chest therapy using the manual method, an a cappella device or a chest physiotherapy vest. Chest physiotherapy can help bring up mucous from the lower bronchial tree, however, an adequate cough is needed to remove secretions. In people who have decreased lung reserve and a weak cough, use of an insufflator-exsufflator (cough-assist) device may be useful as a maintenance therapy or during acute respiratory illnesses to help remove bronchial secretions from the upper airways. Evaluation by a Pulmonology specialist, however, should first be done to properly assess patient suitability.
Children and adults with chronic dry cough, increased work of breathing (fast respiratory rate, shortness of breath at rest or with activities) and absence of an infectious process to explain respiratory symptoms should be evaluated for interstitial lung disease or another intrapulmonary process. Evaluation by a Pulmonologist and a CT scan of the chest should be considered in individuals with symptoms of interstitial lung disease or to rule other non-infectious pulmonary processes. People diagnosed with interstitial lung disease may benefit from systemic steroids.
Feeding, swallowing and nutrition
Oral intake may be aided by teaching persons with A–T how to drink, chew and swallow more safely. The propriety of treatments for swallowing problems should be determined following evaluation by an expert in the field of speech-language pathology. Dieticians may help treat nutrition problems by recommending dietary modifications, including high calorie foods or food supplements.
A feeding (gastrostomy) tube is recommended when any of the following occur:[72]
- A child cannot eat enough to grow or a person of any age cannot eat enough to maintain weight;
- Aspiration is problematic;
- Mealtimes are stressful or too long, interfering with other activities.
Education and socialization
Most children with A–T have difficulty in school because of a delay in response time to visual, verbal or other cues, slurred and quiet speech (dysarthria), abnormalities of eye control (oculomotor apraxia), and impaired fine motor control. Despite these problems, children with A–T often enjoy school if proper accommodations to their disability can be made. The decision about the need for special education classes or extra help in regular classes is highly influenced by the local resources available. Decisions about proper educational placement should be revisited as often as circumstances warrant. Despite their many neurologic impairments, most individuals with A–T are very socially aware and socially skilled, and thus benefit from sustained peer relationships developed at school. Some individuals are able to function quite well despite their disabilities and a few have graduated from community colleges.
Many of the problems encountered will benefit from special attention, as problems are often related more to “input and output” issues than to intellectual impairment. Problems with eye movement control make it difficult for people with A–T to read, yet most fully understand the meaning and nuances of text that is read to them. Delays in speech initiation and lack of facial expression make it seem that they do not know the answers to questions. Reduction of the skilled effort needed to answer questions, and an increase of the time available to respond, is often rewarded by real accomplishment. It is important to recognize that intellectual disability is not regularly a part of the clinical picture of A–T although school performance may be suboptimal because of the many difficulties in reading, writing, and speech. Children with A–T are often very conscious of their appearance, and strive to appear normal to their peers and teachers. Life within the ataxic body can be tiring. The enhanced effort needed to maintain appearances and increased energy expended in abnormal tone and extra movements all contribute to physical and mental fatigue. As a consequence, for some a shortened school day yields real benefits.
General recommendations
- All children with A–T need special attention to the barriers they experience in school. In the United States, this takes the form of a formal IEP (Individualized Education Program).
- Children with A–T tend to be excellent problem solvers. Their involvement in how to best perform tasks should be encouraged.
- Speech-language pathologists may facilitate communication skills that enable persons with A–T to get their messages across (using key words vs. complete sentences) and teach strategies to decrease frustration associated with the increase time needed to respond to questions (e.g., holding up a hand and informing others about the need to allow more time for responses). Rarely helpful are traditional speech therapies that focus on the production of specific sounds and strengthening of the lip and tongue muscles.
- Classroom aides may be appropriate, especially to help with scribing, transportation through the school, mealtimes and toileting. The impact of an aide on peer relationships should be monitored carefully.
- Physical therapy is useful to maintain strength and general cardiovascular health. Horseback therapy and exercises in a swimming pool are often well tolerated and fun for people with A–T. However, no amount of practice will slow the cerebellar degeneration or improve neurologic function. Exercise to the point of exhaustion should be avoided.
- Hearing is normal throughout life. Books on tape may be a useful adjunct to traditional school materials.
- Early use of computers (preschool) with word completion software should be encouraged.
- Practicing coordination (e.g. balance beam or cursive writing exercises) is not helpful.
- Occupational therapy is helpful for managing daily living skills.
- Allow rest time, shortened days, reduced class schedule, reduced homework, modified tests as necessary.
- Like all children, those with A–T need to have goals to experience the satisfaction of making progress.
- Social interactions with peers are important, and should be taken into consideration for class placement. For everyone long-term peer relationships can be the most rewarding part of life; for those with A–T establishing these connections in school years can be helpful.
Treatment
No curative medication has been approved for the treatment of inherited cerebellar ataxias, including Ataxia-Telangiectasia.[73]
N-Acetyl-Leucine
N-Acetyl-Leucine is an orally administered, modified amino acid that is being developed as a novel treatment for multiple rare and common neurological disorders by IntraBio Inc (Oxford, United Kingdom).[74]
N-Acetyl-Leucine has been granted multiple orphan drug designations from the U.S. Food & Drug Administration (FDA)[75] and the European Medicines Agency (EMA)[76] for the treatment of various genetic diseases, including Ataxia-Telangiectasia. N-Acetyl-Leucine has also been granted Orphan Drug Designations in the US and EU for related inherited cerebellar ataxias, such as Spinocerebellar Ataxias. U.S. Food & Drug Administration (FDA)[77] and the European Medicines Agency (EMA).[78]
Published case series studies have demonstrated the positive clinical benefit of treatment with N-Acetyl-Leucine various inherited cerebellar ataxias.[79][80]
A multinational clinical trial investigating N-Acetyl-L-Leucine for the treatment Ataxia-Telangiectasia began in 2019.[81]
IntraBio is also conducting two parallel clinical trials with N-Acetyl-L-Leucine for the treatment Niemann-Pick disease type C[82] and GM2 Gangliosidosis (Tay-Sachs and Sandhoff Disease)[83] Future opportunities to develop N-Acetyl-Leucine include Lewy Body Dementia,[84] Amyotrophic lateral sclerosis, Restless Leg Syndrome, Multiple Sclerosis, and Migraine.[85]
Prognosis
Median survival in two large cohorts studies was 25 and 19 years of age, with a wide range.[86]
Life expectancy does not correlate well with severity of neurological impairment.[86]
Epidemiology
Individuals of all races and ethnicities are affected equally. The incidence worldwide is estimated to be between 1 in 40,000 and 1 in 100,000 people.[5][7]
Research directions
An open-label Phase II clinical trial studying the use of red blood cells (erythrocytes) loaded with dexamethasone sodium phosphate found that this treatment improved symptoms and appeared to be well tolerated.[87] This treatment uses a unique delivery system for medication by using the patient's own red blood cells as the delivery vehicle for the drug.[88]
References
- ↑ Mulliken, John B. (2013). "13. Capillary malformations, hyperkeratotic stains, telangiectasias, and miscellaneous vascular blots". In Mulliken, John B.; Burrows, Patricia E.; Fishman, Steven J. (eds.). Mulliken and Young's Vascular Anomalies: Hemangiomas and Malformations (2nd ed.). Oxford University Press. p. 551. ISBN 978-0-19-972254-9. Archived from the original on 2023-06-30. Retrieved 2023-05-19.
- ↑ Louis-Bar D (1941). "Sur un syndrome progressif cormprenant des télangiectasies capillaires cutanées et conjonctivales symétriques, à disposition naevoïde et des troubles cérébelleux". Confinia Neurologica. 4 (1–2): 32–42. doi:10.1159/000106149.
- ↑ Boder E (1985). "Ataxia–telangiectasia: an overview". Kroc Foundation Series. 19: 1–63. PMID 2415689.
- 1 2 3 Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, Ashkenazi M, Pecker I, Frydman M, Harnik R, Patanjali SR, Simmons A, Clines GA, Sartiel A, Gatti RA, Chessa L, Sanal O, Lavin MF, Jaspers NG, Taylor AM, Arlett CF, Miki T, Weissman SM, Lovett M, Collins FS, Shiloh Y (June 1995). "A single ataxia telangiectasia gene with a product similar to PI-3 kinase". Science. 268 (5218): 1749–53. Bibcode:1995Sci...268.1749S. doi:10.1126/science.7792600. PMID 7792600.
- 1 2 3 Shiloh Y, Kastan MB (2001). "ATM: genome stability, neuronal development, and cancer cross paths". Advances in Cancer Research. 83: 209–54. doi:10.1016/s0065-230x(01)83007-4. ISBN 9780120066834. PMID 11665719.
- ↑ "Ataxia–telangiectasia". Genetics Home Reference. Archived from the original on 2018-03-30. Retrieved 2018-03-29.
- 1 2 Swift M, Morrell D, Cromartie E, Chamberlin AR, Skolnick MH, Bishop DT (November 1986). "The incidence and gene frequency of ataxia–telangiectasia in the United States". American Journal of Human Genetics. 39 (5): 573–83. PMC 1684065. PMID 3788973.
- 1 2 Cabana MD, Crawford TO, Winkelstein JA, Christensen JR, Lederman HM (July 1998). "Consequences of the delayed diagnosis of ataxia–telangiectasia". Pediatrics. 102 (1 Pt 1): 98–100. doi:10.1542/peds.102.1.98. PMID 9651420. S2CID 22538515.
- ↑ Stankovic T, Kidd AM, Sutcliffe A, McGuire GM, Robinson P, Weber P, Bedenham T, Bradwell AR, Easton DF, Lennox GG, Haites N, Byrd PJ, Taylor AM (1998). "ATM mutations and phenotypes in ataxia-telangiectasia families in the British Isles: expression of mutant ATM and the risk of leukemia, lymphoma, and breast cancer". American Journal of Human Genetics. 62 (2): 334–345. doi:10.1086/301706. PMC 1376883. PMID 9463314.
- ↑ Dörk T, Bendix-Waltes R, Wegner RD, Stumm M (2004). "Slow progression of ataxia-telangiectasia with double missense and in frame splice mutations". American Journal of Medical Genetics. 126A (3): 272–277. doi:10.1002/ajmg.a.20601. PMID 15054841. S2CID 22090621.
- ↑ van Os NJ, Chessa L, Weemaes CM, van Deuren M, Fiévet A, van Gaalen J, Mahlaoui N, Roeleveld N, Schrader C, Schindler D, Taylor AM, Van de Warrenburg BP, Dörk T, Willemsen MA (2019). "Genotype-phenotype correlations in ataxia telangiectasia patients with ATM c.3576G>A and c.8147T>C mutations". Journal of Medical Genetics. 56 (5): 308–316. doi:10.1136/jmedgenet-2018-105635. PMID 30819809.
- ↑ Asadollahi R, Britschgi C, Joset P, Oneda B, Schindler D, Meier UR, Rauch A (August 2020). "Severe reaction to radiotherapy provoked by hypomorphic germline mutations in ATM (ataxia-telangiectasia mutated gene)". Molecular Genetics & Genomic Medicine. 8 (10): e1409. doi:10.1002/mgg3.1409. PMC 7549565. PMID 32748564.
- 1 2 Crawford TO (December 1998). "Ataxia telangiectasia". Seminars in Pediatric Neurology. 5 (4): 287–94. doi:10.1016/s1071-9091(98)80007-7. PMID 9874856.
- ↑ Crawford TO, Mandir AS, Lefton-Greif MA, Goodman SN, Goodman BK, Sengul H, Lederman HM (April 2000). "Quantitative neurologic assessment of ataxia–telangiectasia". Neurology. 54 (7): 1505–9. doi:10.1212/wnl.54.7.1505. PMID 10751267. S2CID 24908122.
- ↑ Lavin MF, Gueven N, Bottle S, Gatti RA (2007). "Current and potential therapeutic strategies for the treatment of ataxia–telangiectasia". British Medical Bulletin. 81–82: 129–47. doi:10.1093/bmb/ldm012. PMID 17586848.
- ↑ Lin DD, Barker PB, Lederman HM, Crawford TO (January 2014). "Cerebral abnormalities in adults with ataxia–telangiectasia". AJNR. American Journal of Neuroradiology. 35 (1): 119–23. doi:10.3174/ajnr.A3646. PMC 4106125. PMID 23886747.
- 1 2 Rothblum-Oviatt, Cynthia; Wright, Jennifer; Lefton-Greif, Maureen A.; McGrath-Morrow, Sharon A.; Crawford, Thomas O.; Lederman, Howard M. (2016-11-25). "Ataxia telangiectasia: a review". Orphanet Journal of Rare Diseases. 11 (1): 159. doi:10.1186/s13023-016-0543-7. ISSN 1750-1172. PMC 5123280. PMID 27884168.
- ↑ Nowak-Wegrzyn A, Crawford TO, Winkelstein JA, Carson KA, Lederman HM (April 2004). "Immunodeficiency and infections in ataxia–telangiectasia". The Journal of Pediatrics. 144 (4): 505–11. doi:10.1016/j.jpeds.2003.12.046. PMID 15069401.
- ↑ Reiman A, Srinivasan V, Barone G, Last JI, Wootton LL, Davies EG, Verhagen MM, Willemsen MA, Weemaes CM, Byrd PJ, Izatt L, Easton DF, Thompson DJ, Taylor AM (August 2011). "Lymphoid tumours and breast cancer in ataxia telangiectasia; substantial protective effect of residual ATM kinase activity against childhood tumours". British Journal of Cancer. 105 (4): 586–91. doi:10.1038/bjc.2011.266. PMC 3170966. PMID 21792198.
- ↑ Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, Byrd P, Taylor M, Easton DF (June 2005). "Cancer risks and mortality in heterozygous ATM mutation carriers". Journal of the National Cancer Institute. 97 (11): 813–22. doi:10.1093/jnci/dji141. PMID 15928302.
- ↑ Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N, et al. (Breast Cancer Susceptibility Collaboration, (UK)) (August 2006). "ATM mutations that cause ataxia–telangiectasia are breast cancer susceptibility alleles". Nature Genetics. 38 (8): 873–5. doi:10.1038/ng1837. PMID 16832357. S2CID 2909283.
- ↑ Paller AS, Massey RB, Curtis MA, Pelachyk JM, Dombrowski HC, Leickly FE, Swift M (December 1991). "Cutaneous granulomatous lesions in patients with ataxia–telangiectasia". The Journal of Pediatrics. 119 (6): 917–22. doi:10.1016/s0022-3476(05)83043-4. PMID 1960607.
- 1 2 McGrath-Morrow SA, Gower WA, Rothblum-Oviatt C, Brody AS, Langston C, Fan LL, Lefton-Greif MA, Crawford TO, Troche M, Sandlund JT, Auwaerter PG, Easley B, Loughlin GM, Carroll JL, Lederman HM (September 2010). "Evaluation and management of pulmonary disease in ataxia–telangiectasia". Pediatric Pulmonology. 45 (9): 847–59. doi:10.1002/ppul.21277. PMC 4151879. PMID 20583220.
- ↑ Lefton-Greif MA, Crawford TO, Winkelstein JA, Loughlin GM, Koerner CB, Zahurak M, Lederman HM (February 2000). "Oropharyngeal dysphagia and aspiration in patients with ataxia–telangiectasia". The Journal of Pediatrics. 136 (2): 225–31. doi:10.1016/s0022-3476(00)70106-5. PMID 10657830.
- ↑ Farr AK, Shalev B, Crawford TO, Lederman HM, Winkelstein JA, Repka MX (December 2002). "Ocular manifestations of ataxia–telangiectasia". American Journal of Ophthalmology. 134 (6): 891–6. doi:10.1016/s0002-9394(02)01796-8. PMID 12470759.
- ↑ Gatti RA, Berkel I, Boder E, Braedt G, Charmley P, Concannon P, Ersoy F, Foroud T, Jaspers NG, Lange K (December 1988). "Localization of an ataxia–telangiectasia gene to chromosome 11q22-23". Nature. 336 (6199): 577–80. Bibcode:1988Natur.336..577G. doi:10.1038/336577a0. PMID 3200306. S2CID 4255660.
- 1 2 Derheimer FA, Kastan MB (September 2010). "Multiple roles of ATM in monitoring and maintaining DNA integrity". FEBS Letters. 584 (17): 3675–81. doi:10.1016/j.febslet.2010.05.031. PMC 2950315. PMID 20580718.
- 1 2 Kurz EU, Lees-Miller SP (Aug–Sep 2004). "DNA damage-induced activation of ATM and ATM-dependent signaling pathways". DNA Repair. 3 (8–9): 889–900. doi:10.1016/j.dnarep.2004.03.029. PMID 15279774.
- 1 2 Dar I, Biton S, Shiloh Y, Barzilai A (July 2006). "Analysis of the ataxia telangiectasia mutated-mediated DNA damage response in murine cerebellar neurons". The Journal of Neuroscience. 26 (29): 7767–74. doi:10.1523/JNEUROSCI.2055-06.2006. PMC 6674276. PMID 16855104.
- ↑ Gorodetsky E, Calkins S, Ahn J, Brooks PJ (November 2007). "ATM, the Mre11/Rad50/Nbs1 complex, and topoisomerase I are concentrated in the nucleus of Purkinje neurons in the juvenile human brain". DNA Repair. 6 (11): 1698–707. doi:10.1016/j.dnarep.2007.06.011. PMC 2797317. PMID 17706468.
- 1 2 Valentin-Vega YA, Maclean KH, Tait-Mulder J, Milasta S, Steeves M, Dorsey FC, Cleveland JL, Green DR, Kastan MB (February 2012). "Mitochondrial dysfunction in ataxia–telangiectasia". Blood. 119 (6): 1490–500. doi:10.1182/blood-2011-08-373639. PMC 3286212. PMID 22144182.
- 1 2 Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT (October 2010). "ATM activation by oxidative stress". Science. 330 (6003): 517–21. Bibcode:2010Sci...330..517G. doi:10.1126/science.1192912. hdl:2152/9689. PMID 20966255. S2CID 206527888.
- ↑ Bakkenist CJ, Kastan MB (January 2003). "DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation". Nature. 421 (6922): 499–506. Bibcode:2003Natur.421..499B. doi:10.1038/nature01368. PMID 12556884. S2CID 4403303.
- ↑ Kanu N, Behrens A (November 2008). "ATMINistrating ATM signalling: regulation of ATM by ATMIN". Cell Cycle. 7 (22): 3483–6. doi:10.4161/cc.7.22.7044. PMID 19001856.
- ↑ Shiloh Y (March 2003). "ATM and related protein kinases: safeguarding genome integrity". Nature Reviews. Cancer. 3 (3): 155–68. doi:10.1038/nrc1011. PMID 12612651. S2CID 22770833.
- 1 2 Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A (July 1996). "Atm-deficient mice: a paradigm of ataxia telangiectasia". Cell. 86 (1): 159–71. doi:10.1016/S0092-8674(00)80086-0. PMID 8689683. S2CID 11922073.
- ↑ Plug AW, Peters AH, Xu Y, Keegan KS, Hoekstra MF, Baltimore D, de Boer P, Ashley T (December 1997). "ATM and RPA in meiotic chromosome synapsis and recombination". Nature Genetics. 17 (4): 457–61. doi:10.1038/ng1297-457. PMID 9398850. S2CID 193465.
- ↑ Barchi M, Roig I, Di Giacomo M, de Rooij DG, Keeney S, Jasin M (May 2008). "ATM promotes the obligate XY crossover and both crossover control and chromosome axis integrity on autosomes". PLOS Genetics. 4 (5): e1000076. doi:10.1371/journal.pgen.1000076. PMC 2374915. PMID 18497861.

- ↑ Lumsden JM, McCarty T, Petiniot LK, Shen R, Barlow C, Wynn TA, Morse HC, Gearhart PJ, Wynshaw-Boris A, Max EE, Hodes RJ (November 2004). "Immunoglobulin class switch recombination is impaired in Atm-deficient mice". The Journal of Experimental Medicine. 200 (9): 1111–21. doi:10.1084/jem.20041074. PMC 2211853. PMID 15504820.
- ↑ Franco S, Alt FW, Manis JP (September 2006). "Pathways that suppress programmed DNA breaks from progressing to chromosomal breaks and translocations". DNA Repair. 5 (9–10): 1030–41. doi:10.1016/j.dnarep.2006.05.024. PMID 16934538.
- ↑ Callén E, Jankovic M, Wong N, Zha S, Chen HT, Difilippantonio S, Di Virgilio M, Heidkamp G, Alt FW, Nussenzweig A, Nussenzweig M (May 2009). "Essential role for DNA-PKcs in DNA double-strand break repair and apoptosis in ATM-deficient lymphocytes". Molecular Cell. 34 (3): 285–97. doi:10.1016/j.molcel.2009.04.025. PMC 2709792. PMID 19450527.
- ↑ Bagley J, Singh G, Iacomini J (April 2007). "Regulation of oxidative stress responses by ataxia–telangiectasia mutated is required for T cell proliferation". Journal of Immunology. 178 (8): 4757–63. doi:10.4049/jimmunol.178.8.4757. PMID 17404255.
- ↑ Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, Nuskey B, Sullivan KE, Pandita TK, Bassing CH, Sleckman BP (July 2006). "ATM stabilizes DNA double-strand-break complexes during V(D)J recombination". Nature. 442 (7101): 466–70. Bibcode:2006Natur.442..466B. doi:10.1038/nature04866. PMID 16799570. S2CID 4320129.
- ↑ Bredemeyer AL, Huang CY, Walker LM, Bassing CH, Sleckman BP (August 2008). "Aberrant V(D)J recombination in ataxia telangiectasia mutated-deficient lymphocytes is dependent on nonhomologous DNA end joining". Journal of Immunology. 181 (4): 2620–5. doi:10.4049/jimmunol.181.4.2620. PMC 3598579. PMID 18684952.
- ↑ Bredemeyer AL, Helmink BA, Innes CL, Calderon B, McGinnis LM, Mahowald GK, Gapud EJ, Walker LM, Collins JB, Weaver BK, Mandik-Nayak L, Schreiber RD, Allen PM, May MJ, Paules RS, Bassing CH, Sleckman BP (December 2008). "DNA double-strand breaks activate a multi-functional genetic program in developing lymphocytes". Nature. 456 (7223): 819–23. Bibcode:2008Natur.456..819B. doi:10.1038/nature07392. PMC 2605662. PMID 18849970.
- ↑ Callén E, Jankovic M, Difilippantonio S, Daniel JA, Chen HT, Celeste A, Pellegrini M, McBride K, Wangsa D, Bredemeyer AL, Sleckman BP, Ried T, Nussenzweig M, Nussenzweig A (July 2007). "ATM prevents the persistence and propagation of chromosome breaks in lymphocytes". Cell. 130 (1): 63–75. doi:10.1016/j.cell.2007.06.016. PMID 17599403. S2CID 17514413.
- ↑ Shiloh Y, Tabor E, Becker Y (July 1982). "Colony-forming ability of ataxia–telangiectasia skin fibroblasts is an indicator of their early senescence and increased demand for growth factors". Experimental Cell Research. 140 (1): 191–9. doi:10.1016/0014-4827(82)90169-0. PMID 6213420.
- ↑ Inomata K, Aoto T, Binh NT, Okamoto N, Tanimura S, Wakayama T, Iseki S, Hara E, Masunaga T, Shimizu H, Nishimura EK (June 2009). "Genotoxic stress abrogates renewal of melanocyte stem cells by triggering their differentiation". Cell. 137 (6): 1088–99. doi:10.1016/j.cell.2009.03.037. hdl:2297/19337. PMID 19524511. S2CID 16630060.
- ↑ Stray-Pedersen A, Borresen-Dale AL, Paus E, Lindman CR, Burgers T, Abrahamsen TG (November 2007). "Alpha fetoprotein is increasing with age in ataxia–telangiectasia". European Journal of Paediatric Neurology. 11 (6): 375–80. doi:10.1016/j.ejpn.2007.04.001. PMID 17540590.
- ↑ Biton S, Dar I, Mittelman L, Pereg Y, Barzilai A, Shiloh Y (June 2006). "Nuclear ataxia–telangiectasia mutated (ATM) mediates the cellular response to DNA double strand breaks in human neuron-like cells". The Journal of Biological Chemistry. 281 (25): 17482–91. doi:10.1074/jbc.M601895200. PMID 16627474.
- ↑ Herzog KH, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ (May 1998). "Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system". Science. 280 (5366): 1089–91. Bibcode:1998Sci...280.1089H. doi:10.1126/science.280.5366.1089. PMID 9582124.
- ↑ Lee Y, Chong MJ, McKinnon PJ (September 2001). "Ataxia telangiectasia mutated-dependent apoptosis after genotoxic stress in the developing nervous system is determined by cellular differentiation status". The Journal of Neuroscience. 21 (17): 6687–93. doi:10.1523/JNEUROSCI.21-17-06687.2001. PMC 6763074. PMID 11517258.
- ↑ Sordet O, Redon CE, Guirouilh-Barbat J, Smith S, Solier S, Douarre C, Conti C, Nakamura AJ, Das BB, Nicolas E, Kohn KW, Bonner WM, Pommier Y (August 2009). "Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks". EMBO Reports. 10 (8): 887–93. doi:10.1038/embor.2009.97. PMC 2726680. PMID 19557000.
- ↑ Das BB, Antony S, Gupta S, Dexheimer TS, Redon CE, Garfield S, Shiloh Y, Pommier Y (December 2009). "Optimal function of the DNA repair enzyme TDP1 requires its phosphorylation by ATM and/or DNA-PK". The EMBO Journal. 28 (23): 3667–80. doi:10.1038/emboj.2009.302. PMC 2790489. PMID 19851285.
- ↑ Iourov IY, Vorsanova SG, Liehr T, Kolotii AD, Yurov YB (July 2009). "Increased chromosome instability dramatically disrupts neural genome integrity and mediates cerebellar degeneration in the ataxia–telangiectasia brain". Human Molecular Genetics. 18 (14): 2656–69. doi:10.1093/hmg/ddp207. PMID 19414482.
- ↑ Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, Kusewitt D, Mills GB, Kastan MB, Walker CL (March 2010). "ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS". Proceedings of the National Academy of Sciences of the United States of America. 107 (9): 4153–8. Bibcode:2010PNAS..107.4153A. doi:10.1073/pnas.0913860107. PMC 2840158. PMID 20160076.
- ↑ Cosentino C, Grieco D, Costanzo V (February 2011). "ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair". The EMBO Journal. 30 (3): 546–55. doi:10.1038/emboj.2010.330. PMC 3034007. PMID 21157431.
- ↑ Biton S, Barzilai A, Shiloh Y (July 2008). "The neurological phenotype of ataxia–telangiectasia: solving a persistent puzzle". DNA Repair. 7 (7): 1028–38. doi:10.1016/j.dnarep.2008.03.006. PMID 18456574.
- ↑ D'Souza AD, Parish IA, Krause DS, Kaech SM, Shadel GS (January 2013). "Reducing mitochondrial ROS improves disease-related pathology in a mouse model of ataxia–telangiectasia". Molecular Therapy. 21 (1): 42–8. doi:10.1038/mt.2012.203. PMC 3538311. PMID 23011031.
- ↑ Sharma NK, Lebedeva M, Thomas T, Kovalenko OA, Stumpf JD, Shadel GS, Santos JH (January 2014). "Intrinsic mitochondrial DNA repair defects in Ataxia Telangiectasia". DNA Repair. 13: 22–31. doi:10.1016/j.dnarep.2013.11.002. PMC 6211587. PMID 24342190.
- ↑ Yang Y, Herrup K (March 2005). "Loss of neuronal cell cycle control in ataxia–telangiectasia: a unified disease mechanism". The Journal of Neuroscience. 25 (10): 2522–9. doi:10.1523/JNEUROSCI.4946-04.2005. PMC 6725172. PMID 15758161.
- ↑ Li J, Han YR, Plummer MR, Herrup K (December 2009). "Cytoplasmic ATM in neurons modulates synaptic function". Current Biology. 19 (24): 2091–6. doi:10.1016/j.cub.2009.10.039. PMC 2805770. PMID 19962314.
- ↑ Li J, Chen J, Ricupero CL, Hart RP, Schwartz MS, Kusnecov A, Herrup K (May 2012). "Nuclear accumulation of HDAC4 in ATM deficiency promotes neurodegeneration in ataxia telangiectasia". Nature Medicine. 18 (5): 783–90. doi:10.1038/nm.2709. PMC 3378917. PMID 22466704.
- ↑ Herrup K (October 2013). "ATM and the epigenetics of the neuronal genome". Mechanisms of Ageing and Development. 134 (10): 434–9. doi:10.1016/j.mad.2013.05.005. PMC 3791148. PMID 23707635.
- ↑ Li J, Hart RP, Mallimo EM, Swerdel MR, Kusnecov AW, Herrup K (December 2013). "EZH2-mediated H3K27 trimethylation mediates neurodegeneration in ataxia–telangiectasia". Nature Neuroscience. 16 (12): 1745–53. doi:10.1038/nn.3564. PMC 3965909. PMID 24162653.
- ↑ Jiang D, Zhang Y, Hart RP, Chen J, Herrup K, Li J (December 2015). "Alteration in 5-hydroxymethylcytosine-mediated epigenetic regulation leads to Purkinje cell vulnerability in ATM deficiency". Brain. 138 (Pt 12): 3520–36. doi:10.1093/brain/awv284. PMC 4668921. PMID 26510954.
- ↑ Wood LM, Sankar S, Reed RE, Haas AL, Liu LF, McKinnon P, Desai SD (January 2011). "A novel role for ATM in regulating proteasome-mediated protein degradation through suppression of the ISG15 conjugation pathway". PLOS ONE. 6 (1): e16422. Bibcode:2011PLoSO...616422W. doi:10.1371/journal.pone.0016422. PMC 3027683. PMID 21298066.
- ↑ Sun X, Becker-Catania SG, Chun HH, Hwang MJ, Huo Y, Wang Z, Mitui M, Sanal O, Chessa L, Crandall B, Gatti RA (June 2002). "Early diagnosis of ataxia–telangiectasia using radiosensitivity testing". The Journal of Pediatrics. 140 (6): 724–31. doi:10.1067/mpd.2002.123879. PMID 12072877.
- ↑ Chun HH, Sun X, Nahas SA, Teraoka S, Lai CH, Concannon P, Gatti RA (December 2003). "Improved diagnostic testing for ataxia–telangiectasia by immunoblotting of nuclear lysates for ATM protein expression". Molecular Genetics and Metabolism. 80 (4): 437–43. doi:10.1016/j.ymgme.2003.09.008. PMID 14654357.
- ↑ Taylor AM, Byrd PJ (October 2005). "Molecular pathology of ataxia telangiectasia". Journal of Clinical Pathology. 58 (10): 1009–15. doi:10.1136/jcp.2005.026062. PMC 1770730. PMID 16189143.
- ↑ Anheim M, Tranchant C, Koenig M (February 2012). "The autosomal recessive cerebellar ataxias". The New England Journal of Medicine. 366 (7): 636–46. doi:10.1056/NEJMra1006610. hdl:10068/785252. PMID 22335741.
- ↑ Lefton-Greif MA, Crawford TO, McGrath-Morrow S, Carson KA, Lederman HM (May 2011). "Safety and caregiver satisfaction with gastrostomy in patients with Ataxia Telangiectasia". Orphanet Journal of Rare Diseases. 6: 23. doi:10.1186/1750-1172-6-23. PMC 3116459. PMID 21569628.
- ↑ Ilg W, Bastian AJ, Boesch S, Burciu RG, Celnik P, Claaßen J, et al. (April 2014). "Consensus paper: management of degenerative cerebellar disorders". Cerebellum. 13 (2): 248–68. doi:10.1007/s12311-013-0531-6. PMC 4344126. PMID 24222635.
- ↑ "IntraBio". Archived from the original on 2019-08-01. Retrieved 2019-08-01.
- ↑ "Search Orphan Drug Designations and Approvals". www.accessdata.fda.gov. Archived from the original on 2021-04-27. Retrieved 2019-08-01.
- ↑ "Search Orphan Drug Designations and Approvals". www.accessdata.fda.gov. Archived from the original on 2021-04-28. Retrieved 2019-08-01.
- ↑ "Search Orphan Drug Designations and Approvals". www.accessdata.fda.gov. Archived from the original on 2021-04-28. Retrieved 2019-08-01.
- ↑ FRANCISCO, Estela Miranda (2018-12-20). "EU/3/18/2059". European Medicines Agency. Archived from the original on 2019-08-01. Retrieved 2019-08-01.
- ↑ Strupp M, Teufel J, Habs M, Feuerecker R, Muth C, van de Warrenburg BP, et al. (October 2013). "Effects of acetyl-DL-leucine in patients with cerebellar ataxia: a case series". Journal of Neurology. 260 (10): 2556–61. doi:10.1007/s00415-013-7016-x. PMC 3824630. PMID 23835634.
- ↑ Schniepp R, Strupp M, Wuehr M, Jahn K, Dieterich M, Brandt T, Feil K (2016). "Acetyl-DL-leucine improves gait variability in patients with cerebellar ataxia-a case series". Cerebellum & Ataxias. 3: 8. doi:10.1186/s40673-016-0046-2. PMC 4828858. PMID 27073690.
- ↑ Clinical trial number NCT03759678 for "N-Acetyl-L-Leucine for Ataxia-Telangiectasia (A-T)" at ClinicalTrials.gov
- ↑ Clinical trial number NCT03759639 for "N-Acetyl-L-Leucine for Niemann-Pick Disease, Type C (NPC)" at ClinicalTrials.gov
- ↑ Clinical trial number NCT03759665 for "N-Acetyl-L-Leucine for GM2 Gangliosdisosis (Tay-Sachs and Sandhoff Disease)" at ClinicalTrials.gov
- ↑ "IntraBio". Archived from the original on 2019-08-01. Retrieved 2019-08-01.
- ↑ Strupp M, Bayer O, Feil K, Straube A (February 2019). "Prophylactic treatment of migraine with and without aura with acetyl-DL-leucine: a case series". Journal of Neurology. 266 (2): 525–529. doi:10.1007/s00415-018-9155-6. PMID 30547273. S2CID 56148131.
- 1 2 Crawford, T O (2005). "Survival probability in ataxia telangiectasia". Archives of Disease in Childhood. 91 (7): 610–611. doi:10.1136/adc.2006.094268. ISSN 0003-9888. PMC 2082822. PMID 16790721.
- ↑ Chessa L, Leuzzi V, Plebani A, Soresina A, Micheli R, D'Agnano D, Venturi T, Molinaro A, Fazzi E, Marini M, Ferremi Leali P, Quinti I, Cavaliere FM, Girelli G, Pietrogrande MC, Finocchi A, Tabolli S, Abeni D, Magnani M (January 2014). "Intra-erythrocyte infusion of dexamethasone reduces neurological symptoms in ataxia teleangiectasia patients: results of a phase 2 trial". Orphanet Journal of Rare Diseases. 9 (1): 5. doi:10.1186/1750-1172-9-5. PMC 3904207. PMID 24405665.
- ↑ Yousefpour P, Chilkoti A (September 2014). "Co-opting biology to deliver drugs". Biotechnology and Bioengineering. 111 (9): 1699–716. doi:10.1002/bit.25307. PMC 4251460. PMID 24916780.
External links
- About A–T from the NINDS Archived 2016-12-14 at the Wayback Machine
- Orphanet for A–T Archived 2021-05-27 at the Wayback Machine
- GeneReviews for ataxia–telangiectasia Archived 2009-01-17 at the Wayback Machine
- Replication-Independent Double-Strand Breaks (DSBs) Archived 2009-02-21 at the Wayback Machine Discusses importance of the ATM kinase
| Classification | |
|---|---|
| External resources |
|