Neurofibromatosis
| Neurofibromatosis | |
|---|---|
 | |
| Back of a woman with neurofibromatosis | |
| Specialty | Neurosurgery |
| Symptoms | Small lumps within the skin, scoliosis, hearing loss, vision loss[1] |
| Usual onset | Birth to early adulthood[1] |
| Duration | Life long[1] |
| Types | |
| Causes | Genetic[1] |
| Diagnostic method | Symptoms, genetic testing[3] |
| Treatment | Surgery, radiation therapy[3] |
| Prognosis | NF1: normal life expectancy[1] NF2: shortened life expectancy[1] |
| Frequency | 1 in 3,000 people (United States)[1] |
Neurofibromatosis (NF) is a group of four conditions in which tumors grow in the nervous system, bones and skin.[4] The types are neurofibromatosis type I (NF1), neurofibromatosis type II (NF2), schwannomatosis, and neurofibromatosis type 4.[1][2] In NF1 symptoms include light brown spots on the skin and colored part of the eye, freckles in the armpit and groin, small bumps within nerves, and scoliosis.[3] In NF2, there may be hearing loss, cataracts at a young age, balance problems, flesh colored skin flaps, and muscle wasting.[3] In schwannomatosis there may be pain either in one location or in wide areas of the body.[5] The tumors in NF are generally non-cancerous.[1]
The cause is a genetic mutation in certain genes.[1] These can be inherited from a person's parents, or in about half of cases spontaneously occur during early development.[1] Different mutations result in the three types of NF.[6] The tumors involve the supporting cells of the nervous system rather than the neurons.[1] In NF1 the tumors are neurofibromas (tumors of the peripheral nerves), while in NF2 and schwannomatosis tumors of Schwann cells are more common.[1] Diagnosis is typically based on symptoms, examination, medical imaging, and biopsy.[7][5] Genetic testing may rarely be done to support the diagnosis.[3]
There is no known prevention or cure.[1][3] Surgery may be done to remove tumors that are causing problems or have become cancerous.[1] Radiation and chemotherapy may also be used if cancer occurs.[1] A cochlear implant or auditory brainstem implant may help some who have hearing loss due to the condition.[1]
In the United States, about 1 in 3,500 people have NF1 and 1 in 25,000 have NF2.[1] Males and females are affected equally often.[3] In NF1, symptoms are often present at birth or develop before 10 years of age.[1] While the condition typically worsens with time, most people with NF1 have a normal life expectancy.[1] In NF2, symptoms may not become apparent until early adulthood.[1] NF2 increases the risk of early death.[1] Descriptions of the condition occur as far back as the 1st century.[8] It was formally described by Friedrich Daniel von Recklinghausen in 1882, after whom it was previously named.[6]
Signs and symptoms
Neurofibromatosis type 1 in early life may cause learning and behavior problems – about 60% of children who have NF1 have mild difficulty in school.[9] Signs the individual might have are as follows:[10][11]
- Six or more light brown dermatological spots ("café au lait spots")
- At least two neurofibromas
- At least two growths on the eye's iris
- Abnormal growth of the spine (scoliosis)
People with neurofibromatosis type 2 can exhibit the same type of skin symptoms as type 1, but not necessarily in every case.[12] The symptom most characteristic of NF2 is hearing loss. The hearing loss occurs due to the pressure of tumors on the acoustic nerve. The same pressure can cause headaches, dizziness, and nausea.[12]
The main symptom of schwannomatosis is localized pain. This pain is due to tissues and nerves experiencing more pressure because of nearby tumors.[13]
.jpg.webp) Neurofibromatosis
Neurofibromatosis.jpg.webp) Neurofibromatosis
Neurofibromatosis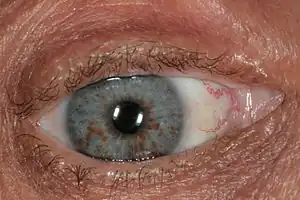 Lisch nodules as seen in NF1
Lisch nodules as seen in NF1
Cause
The three types of Neurofibromatosis are caused by different mutations on chromosomes. NF1 is caused by a mutation on the NF1 gene on the arm of chromosome 17.[14] NF2 is caused by a mutation on the NF2 tumor suppressor gene on chromosome 22.[14] Schwannomatosis is caused by various mutations on chromosome 22.[14]
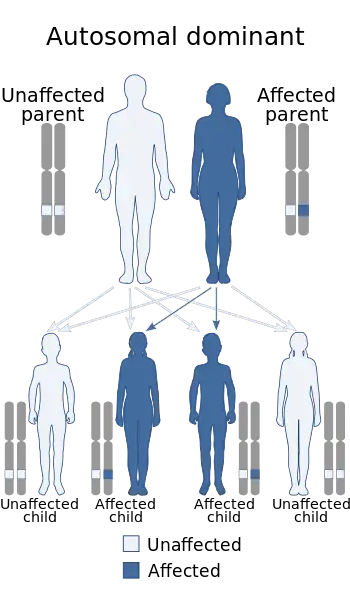
Neurofibromatosis is an autosomal dominant disorder, which means only one copy of the affected gene is needed for the disorder to develop.[15] If one parent has neurofibromatosis, his or her children have a 50% chance of developing the condition as well. The severity of the parent's condition does not affect the child; the affected child may have mild NF1 even though it was inherited from a parent with a severe form of the disorder.[16] The types of neurofibromatosis are:
- Neurofibromatosis type I, in which the nerve tissue grows tumors (neurofibromas) that may be benign, but may cause serious damage by compressing nerves and other tissues.[17]
- Neurofibromatosis type II, in which bilateral acoustic neuromas (tumors of the vestibulocochlear nerve or cranial nerve 8 (CN VIII) also known as schwannoma) develop, often leading to hearing loss.[18]
- Schwannomatosis, in which painful schwannomas develop on spinal and peripheral nerves.[19]
Pathophysiology
Neurofibromatosis type I is caused by a mutation on chromosome 17 encoding a cytoplasmic protein known as neurofibromin.[20] This protein is a tumor suppressor and therefore serves as a signal regulator of cell proliferation and differentiation. A dysfunction or lack of neurofibromin can affect regulation, and cause uncontrolled cell proliferation, leading to the tumors (neurofibromas) that characterize NF1. The neurofibromas caused by NF consist of Schwann cells, fibroblasts, perineuronal cells, mast cells and axons embedded in an extracellular matrix.[21][22] Another function of neurofibromin is to bind to microtubules that play a role in the release of adenylyl cyclase and its activity.[21] Adenylyl cyclase plays an essential role in cognition.[21] Neurofibromin's role in the activity of adenylyl cyclase explains why patients with NF experience cognitive impairment.[21]
Neurofibromatosis type II is caused by a mutation on chromosome 22.[23] The mutation falls on the NF2 tumor suppressor gene.[23] The gene normally encodes a cytoplasmic protein known as merlin. The normal function of merlin is to regulate the activity of multiple growth factors, the mutated copy of the gene leads to merlin's loss of function.[23] The loss of function leads to increased activity of growth factors normally regulated by merlin, leading to the formation of the tumors associated with NF2.[23]
Schwannomatosis is caused by a mutation on the SMARCB1 gene.[13] This gene is located near the NF2 tumor suppressor gene leading to the thought that schwannomatosis and NF2 were the same condition. The two conditions show different mutations on two different genes. The normal function of the SMARCB1 gene is to encode a protein called SMARCB1 that is part of a larger protein complex whose function is not completely understood.[13] The complex including SMARCB1 plays a role in tumor suppression.[13] The mutation of the SMARCB1 gene causes a loss of function in the complex leading to the formation of tumors indicative of schwannomatosis.[13]
Diagnosis
The neurofibromatoses are considered as RASopathies and as members of the neurocutaneous syndromes (phakomatoses).[24] The diagnosis of neurofibromatosis is done via the following means:[25]
- Radiograph
- MRI or CT scan
- EEG
- Slit-lamp examination
- Genetic testing
- Histology
Differential diagnosis
Conditions similar to NF include:
Treatment
Surgical removal of tumors is an option; however, the risks involved should be assessed first.[29] With regard to OPG (optic pathway gliomas), the preferred treatment is chemotherapy. However, radiotherapy is not recommended in children who present with this disorder.[30] It is recommended that children diagnosed with NF1 at an early age have an examination each year, which allows any potential growths or changes related to the disorder to be monitored.[31]
Prognosis
In most cases, symptoms of NF1 are mild, and individuals live normal and productive lives. In some cases, however, NF1 can be severely debilitating and may cause cosmetic and psychological issues. The course of NF2 varies greatly among individuals. In some cases of NF2, the damage to nearby vital structures, such as other cranial nerves and the brain stem, can be life-threatening. Most individuals with schwannomatosis have significant pain. In some extreme cases the pain will be severe and disabling.[11]
Epidemiology
In the United States, about 1 in 3,500 people have NF1, 1 in 25,000 have NF2, and 1 in 40,000 have schwannomatosis.[1] Males and females are affected equally often in all three conditions.[3] In NF1, symptoms are often present at birth or develop before 10 years of age.[1] While the condition typically worsens with time, most people with NF1 have a normal life expectancy.[1] In NF2, symptoms may not become apparent until early adulthood.[1] NF2 increases the risk of early death.[1] Schwannomatosis symptoms develop in early childhood and can worsen with time. Typically life expectancy is unaffected in those with schwannomatosis.[5]
History
Descriptions of what is believed to be the condition go back as far back as the 1st century.[8] The conditions were formally described by Friedrich Daniel von Recklinghausen in 1882, after whom it was previously named.[6]
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 "Neurofibromatosis Fact Sheet". NINDS. 3 February 2016. Archived from the original on 23 January 2018. Retrieved 16 April 2018.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - 1 2 "Neurofibromatosis, type IV, of Riccardi (Concept Id: C0220695) - MedGen - NCBI". www.ncbi.nlm.nih.gov. Archived from the original on 10 July 2019. Retrieved 17 October 2021.
- 1 2 3 4 5 6 7 8 "Learning about Neurofibromatosis". National Human Genome Research Institute (NHGRI). 16 August 2016. Archived from the original on 10 October 2016. Retrieved 7 November 2016. This article incorporates text from this source, which is in the public domain.
- ↑ James, William D.; Elston, Dirk; Treat, James R.; Rosenbach, Misha A.; Neuhaus, Isaac (2020). "27. Genodermatoses and congenital anomalies". Andrews' Diseases of the Skin: Clinical Dermatology (13th ed.). Elsevier. p. 550-553. ISBN 978-0-323-54753-6. Archived from the original on 17 October 2021. Retrieved 17 October 2021.
- 1 2 3 Dhamija, Radhika; Plotkin, Scott; Asthagiri, Ashok; Messiaen, Ludwine; Babovic-Vuksanovic, Dusica (1993). "Schwannomatosis". GeneReviews®. University of Washington, Seattle. PMID 29517885. Archived from the original on 13 August 2020. Retrieved 21 November 2019.
- 1 2 3 Woodrow, Christopher; Clarke, Anna; Amirfeyz, Rouin (1 June 2015). "Neurofibromatosis". Orthopaedics and Trauma. 29 (3): 206–210. doi:10.1016/j.mporth.2015.02.004. ISSN 1877-1327. Archived from the original on 28 August 2021. Retrieved 22 November 2019.
- ↑ Le, C; Bedocs, PM (January 2019). "Neurofibromatosis". PMID 29083784.
{{cite journal}}: Cite journal requires|journal=(help) - 1 2 Evans, Rosalie E. Ferner, Susan M. Huson, D. Gareth R. (2011). Neurofibromatoses in clinical practice. London: Springer. p. 1. ISBN 978-0-85729-628-3. Archived from the original on 10 September 2017. Retrieved 9 October 2015.
- ↑ "Neurofibromatosis". NHS Choices. NHS. Archived from the original on 25 September 2015. Retrieved 9 October 2015.
- ↑ "Neurofibromatosis". NINDS. NIH. Archived from the original on 4 October 2015. Retrieved 9 October 2015.
- 1 2 "NINDS Neurofibromatosis Information Page". 23 February 2015. Archived from the original on 4 April 2015. Retrieved 2015-04-21.
- 1 2 Guha, Martin (2011-03-29). "The Gale Encyclopedia of Genetic Disorders (3rd ed.)2011133Edited by Laurie J. Fundukian. The Gale Encyclopedia of Genetic Disorders (3rd ed.). Detroit, MI: Gale 2010. , ISBN 978 1 4144 7602 5 (print); 978 1 4144 7605 6 (e‐book) $445 (print) 3 vols". Reference Reviews. 25 (3): 40–42. doi:10.1108/09504121111119022. ISSN 0950-4125.
- 1 2 3 4 5 Plotkin, Scott R.; Blakeley, Jaishri O.; Evans, D. Gareth; Hanemann, C. Oliver; Hulsebos, Theo J.M.; Hunter-Schaedle, Kim; Kalpana, Ganjam V.; Korf, Bruce; Messiaen, Ludwine; Papi, Laura; Ratner, Nancy (2013). "Update from the 2011 International Schwannomatosis Workshop: From genetics to diagnostic criteria". American Journal of Medical Genetics Part A. 161 (3): 405–416. doi:10.1002/ajmg.a.35760. PMC 4020435. PMID 23401320.
- 1 2 3 Woodrow, Christopher; Clarke, Anna; Amirfeyz, Rouin (2015). "Neurofibromatosis". Orthopaedics and Trauma. 29 (3): 206–210. doi:10.1016/j.mporth.2015.02.004.
- ↑ Woodrow, Christopher; Clarke, Anna; Amirfeyz, Rouin (2015-06-01). "Neurofibromatosis". Orthopaedics and Trauma. 29 (3): 206–210. doi:10.1016/j.mporth.2015.02.004. ISSN 1877-1327. Archived from the original on 28 August 2021. Retrieved 26 September 2019.
- ↑ Choices, NHS. "Neurofibromatosis type 1 - Causes - NHS Choices". www.nhs.uk. Archived from the original on 24 September 2015. Retrieved 2015-10-09.
- ↑ "Neurofibromatosis type 1". Genetics Home Reference. 5 October 2015. Archived from the original on 10 September 2015. Retrieved 2015-10-09.
- ↑ "Neurofibromatosis type 2". Genetics Home Reference. 5 October 2015. Archived from the original on 10 September 2015. Retrieved 2015-10-09.
- ↑ Perry, Arie; Brat, Daniel J. (1 January 2010). Practical Surgical Neuropathology: A Diagnostic Approach. Elsevier Health Sciences. p. 435. ISBN 978-0443069826. Archived from the original on 2 May 2016.
- ↑ "Orphanet: Neurofibromatosis type 1". www.orpha.net. Archived from the original on 6 October 2015. Retrieved 2015-10-13.
- 1 2 3 4 Ferner, Rosalie E. (2007). "Neurofibromatosis 1". European Journal of Human Genetics. 15 (2): 131–138. doi:10.1038/sj.ejhg.5201676. ISSN 1476-5438. PMID 16957683.
- ↑ Boyd, Kevin P.; Korf, Bruce R.; Theos, Amy (July 2009). "Neurofibromatosis type 1". Journal of the American Academy of Dermatology. 61 (1): 1–14. doi:10.1016/j.jaad.2008.12.051. PMC 2716546. PMID 19539839.
- 1 2 3 4 Lin, Andrew (2013). "Advances in the treatment of neurofibromatosis-associated tumours". Nature Reviews Clinical Oncology. 10 (11): 616–24. doi:10.1038/nrclinonc.2013.144. PMID 23939548. Archived from the original on 28 August 2021. Retrieved 21 November 2019 – via Gale.
- ↑ Conrad Fischer; Farshad Bagheri; Rajpal Manchandani; Richard Pinsker; Sudheer Chauhan; Parenkumar Patel; Mohammad Maruf; Dhaval Satani; Kaushik Doshi; Ayaz Alwani; Naveen Pathak; Craigh Thurm; Mohammad Babury; Mahendra C. Patel; Arthur Shalanov; Samir Sarkar; Sabiha Raouf; Jebun Nahar; Prakashkumar Patel (2010). Master the Board USMLE Step 2 CK. KAPLAN Medical. p. 287. ISBN 978-1-60714-653-7.
- ↑ "Neurofibromatosis. What is neurofibromatosis? Type 1 (NF1) | Patient". Patient. Archived from the original on 4 October 2015. Retrieved 2015-10-09.
- ↑ Friedman, J. M. (2014). Pagon, Roberta A.; Adam, Margaret P.; Ardinger, Holly H.; Wallace, Stephanie E.; Amemiya, Anne; Bean, Lora JH; Bird, Thomas D.; Dolan, Cynthia R.; Fong, Chin-To (eds.). Neurofibromatosis 1. Seattle (WA): University of Washington, Seattle. PMID 20301288. Archived from the original on 18 January 2017.
- ↑ Stevenson, David; Viskochil, David; Mao, Rong (2015). Pagon, Roberta A.; Adam, Margaret P.; Ardinger, Holly H.; Wallace, Stephanie E.; Amemiya, Anne; Bean, Lora JH; Bird, Thomas D.; Dolan, Cynthia R.; Fong, Chin-To (eds.). Legius Syndrome. Seattle (WA): University of Washington, Seattle. PMID 20945555. Archived from the original on 10 September 2017.
- ↑ El-Sobky, Tamer Ahmed; Elsayed, Solaf M.; El Mikkawy, Dalia M.E. (2015). "Orthopaedic manifestations of Proteus syndrome in a child with literature update". Bone Reports. 3: 104–108. doi:10.1016/j.bonr.2015.09.004. PMC 5365241. PMID 28377973.
- ↑ Choices, NHS. "Neurofibromatosis type 2 - Treatment - NHS Choices". www.nhs.uk. Archived from the original on 22 December 2015. Retrieved 2015-10-11.
- ↑ "Complex Neufibrmatosis type 1" (PDF). NHS.uk. NHS. Archived (PDF) from the original on 23 December 2015. Retrieved 13 October 2015.
- ↑ Choices, NHS. "Neurofibromatosis type 1 - Treatment - NHS Choices". www.nhs.uk. Archived from the original on 26 September 2015. Retrieved 2015-10-11.
External links
- National Institute of Neurological Disorders and Stroke - information on Neurofibromatosis Archived 16 August 2019 at the Wayback Machine
- Upadhyaya, Meena; Cooper, David (29 January 2013). Neurofibromatosis Type 1: Molecular and Cellular Biology. Springer Science & Business Media. ISBN 9783642328640. Archived from the original on 23 April 2016. Retrieved 9 October 2015.
| Classification | |
|---|---|
| External resources |