Dravet syndrome
| Dravet syndrome | |
|---|---|
| Other names: Severe myoclonic epilepsy of infancy (SMEI); severe myoclonic epilepsy of infancy borderland (SMEB);[1] polymorphic epilepsy of infancy (PMEI)[2] | |
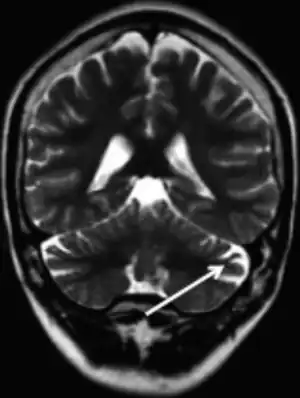 | |
| MRI (adult) showing cerebellar atrophy in Dravet syndrome | |
| Pronunciation |
|
| Specialty | Neurology |
| Symptoms | Prolonged seizures (status epilepticus) triggered by hot temperatures[3][2] |
| Complications | Intellectual dysfunction, behavioral problems[1] |
| Usual onset | First year of life[1] |
| Causes | Genetic mutation in the SCN1A gene[4] |
| Diagnostic method | Based on symptoms and genetic testing[5] |
| Differential diagnosis | Lennox-Gastaut syndrome, epilepsy with myoclonic-atonic seizures, generalized epilepsy with febrile seizures plus[6] |
| Treatment | Anticonvulsant medication, ketogenic diet[2] |
| Medication | Clobazam, fenfluramine, cannabidiol[2][4] |
| Frequency | 1 in 16,000 people[2] |
| Deaths | Up to 20% die by adulthood[2] |
Dravet syndrome, previously known as severe myoclonic epilepsy of infancy (SMEI), is a form of epilepsy that generally begins in the first year of life.[1] Symptoms include prolonged seizures (status epilepticus) that are often triggered by hot temperatures, fever, or visual effects.[3][7][2] Initially seizures may be focal or generalized.[4] Complications may include intellectual dysfunction and behavioral problems.[1]
The cause if often a genetic mutation in the SCN1A gene.[4] Around 90% of mutations newly occur during early development, rather then being inherited from a person's parents.[3] A family history of seizures is present in nearly half of cases.[8] Diagnosis is based on symptoms and genetic testing.[5] MRI of the brain is typically normal initially, though may show changes such as atrophy in some adults.[2]
Treatment is with anticonvulsant medications, such as clobazam.[2] Fenfluramine and cannabidiol have specifically been approved for the condition.[4] A ketogenic diet may also help.[2] Up to 20% of people die by adulthood.[2]
Dravet syndrome occurs in about 1 in 16,000 people.[2] It is the underlying cause of about 2 in 1,000 cases of epilepsy.[2] Males are affected about twice as often as females.[9] The condition was first described in 1978 by Charlotte Dravet.[10]
Signs and symptoms
Dravet syndrome has been characterized by prolonged febrile and non-febrile seizures within the first year of a child's life. This disease progresses to other seizure types like myoclonic and partial seizures, psychomotor delay, and ataxia.[11] It is characterized by cognitive impairment, behavioural disorders, and motor deficits.[12] Behavioural deficits often include hyperactivity and impulsiveness, and in more rare cases, autistic-like behaviours.[12] Dravet syndrome is also associated with sleep disorders including somnolence and insomnia.[12] The seizures experienced by people with Dravet syndrome become worse as the patient ages, as the disease is not very observable when symptoms first appear.[12] This coupled with the range of severity differing between each individual diagnosed and the resistance of these seizures to drugs has made it challenging to develop treatments.[12]
Dravet syndrome appears during the first year of life, often beginning around six months of age with frequent febrile seizures (fever-related seizures). Children with Dravet syndrome typically experience a lagged development of language and motor skills, hyperactivity and sleep difficulties, chronic infection, growth and balance issues, and difficulty relating to others. The effects of this disorder do not diminish over time, and children diagnosed with Dravet syndrome require fully committed caretakers with tremendous patience and the ability to closely monitor them.[13]
Febrile seizures are divided into two categories known as simple and complex. A febrile seizure would be categorized as complex if it has occurred within 24 hours of another seizure or if it lasts longer than 15 minutes. A febrile seizure lasting less than 15 minutes would be considered simple. Sometimes modest hyperthermic stressors like physical exertion or a hot bath can provoke seizures in affected individuals.[13] However, any seizure uninterrupted after 5 minutes, without a resumption of postictal (more normal; recovery-type; after-seizure) consciousness can lead to potentially fatal status epilepticus.
Complications
Although it is not clear whether people with Dravet syndrome are specially vulnerable to COVID-19 infection, recent publications have shown that affected individuals and their families have suffered some indirect damages during COVID-19 pandemic, such as healthcare barriers, loss of therapies or economic issues.[14]
Causes
In most cases the mutations in Dravet syndrome are not hereditary and the mutated gene is found for the first time in a single family member.[11] In 70–90% of patients, Dravet syndrome is caused by nonsense mutations in the SCN1A gene resulting in a premature stop codon and thus a non-functional protein.[11] This gene normally codes for neuronal voltage-gated sodium channel Nav1.1.[15] In mouse models, these loss-of-function mutations have been observed to result in a decrease in sodium currents and impaired excitability of GABAergic interneurons of the hippocampus.[15] The researchers found that loss of Nav1.1 channels was sufficient to cause the epilepsy and premature death seen in Dravet syndrome.[15][16]
The timing of the first signs and symptoms in Dravet syndrome occur about the same time as normal childhood vaccinations, leading some to believe the vaccine was the cause. However, this is likely a non-specific response to fever, as vaccination often induces fever,[17] and fever is known to be associated with seizures in persons with Dravet syndrome.[18] Some of the patients who put forth vaccine injury claims from encephalopathy were later found, upon testing, to actually have Dravet syndrome.[19]
Genetics
The genotypic explanation of the disorder has been located on the specific voltage-gated sodium channel genes known as SCN1A and SCN2A. These genes are located on the long (q) arm of chromosome 2 at position 24.3 and code for the alpha subunit of the transmembrane sodium channel protein. A mutation in either of these two genes will cause an individual to develop dysfunctional sodium channels, which are crucial in the pathway for sending chemical signals in the brain, causing the phenotypic display of myoclonic epilepsy from the individual. A properly functioning channel would respond to a voltage difference across the membrane and form a pore through which only sodium ions can pass. The influx of sodium induces the generation of action potential by temporarily changing the charge of the cell. When the gene is mutated, the eventually translated protein improperly folds its pore segment within the cell membrane because it has different amino acid chemistry, which renders the channel inactive. It is also possible for a mutation to reduce the number of channels produced by an individual, which leads to the development of Dravet syndrome.[20]
Currently, the SCN1A gene is the most clinically relevant; the largest number of epilepsy related mutations characterized thus far occur in this gene.[13][21] Typically, a missense mutation in either the S5 or S6 segment of the sodium channel pore results in a loss of channel function and the development of Dravet syndrome. A heterozygous inheritance of an SCN1A mutation is all that is necessary to develop a defective sodium channel; patients with Dravet syndrome will still have one normal copy of the gene.[20]
Diagnosis
According to the Dravet Syndrome Foundation, the diagnostic criteria for DS requires the patient to present with several of the following symptoms:[22]
- Onset of seizures in the first year of life in an otherwise healthy infant
- Initial seizures are typically prolonged and are generalized or unilateral
- Presence of other seizure types (i.e. myoclonic seizures)
- Seizures associated with fever due to illness or vaccinations
- Seizures induced by prolonged exposure to warm temperatures
- Seizures in response to strong lighting or certain visual patterns
- Initially normal EEGs and later EEGs with slowing and severe generalized polyspikes
- Normal initial development followed by slow development during the first few years of life
- Some degree of hypotonia
- Unstable gait and balance issues
- Ankle pronation and flat feet and/or development of a crouched gait with age
Treatment
Seizures in Dravet syndrome can be difficult to manage but may be reduced by anticonvulsant medications such as clobazam, stiripentol, topiramate and valproate.[23] Because the course of the disorder varies from individual to individual, treatment protocols may vary. A diet high in fats and low in carbohydrates may also be beneficial, known as a ketogenic diet. Although diet adjustment can help, it does not eliminate the symptoms. Until a better form of treatment or cure is discovered, those with this disease will have myoclonic epilepsy for the rest of their lives.[13]
Certain anticonvulsant medications that are classed as sodium channel blockers are now known to make seizures worse in most Dravet patients. These medications include carbamazepine, gabapentin, lamotrigine, and phenytoin.[24]
Treatments include cognitive rehabilitation through psychomotor and speech therapy.[12] In addition, valproate is often administered to prevent recurrence of febrile seizures and a benzodiazepine is used for long lasting seizures, but these treatments are usually insufficient.[25]
Stiripentol was the only medication for which a double-blind placebo-controlled randomized controlled trial was performed and this medication showed efficacy in trials.[25] It acts as a GABAergic agent and as a positive allosteric modulator of GABAA receptor.[25] Stiripentol, can improve focal refractory epilepsy, as well as Dravet's syndrome, supplemented with clobazam and valproate was approved in Europe in 2007 as a therapy for Dravet syndrome and has been found to reduce overall seizure rate by 70%.[25] In cases with more drug-resistant seizures, topiramate and the ketogenic diet are used as alternative treatments.[25][26]
Cannabidiol (CBD) was approved in United States for treatment of Dravet syndrome in 2018.[27] A 2017 study showed that the frequency of seizures per month decreased from 12 to 6 with the use of cannabidiol, compared with a decrease from 15 to 14 with placebo.[28]
In 2020, fenfluramine was approved for the medical treatment in the European Union and the USA.[29][30][31]
Adults
Regarding the care of adult, no particular guidelines are available. The usage of VPA, CLB, and TPM continued through childhood, adolescence, and adulthood, although that of STP decreased with age (31% in adults), according to the findings of a survey of caretakers of patients with DS on their experiences with management and health services.6 According to reports, only a tiny percentage of adult patients receive treatment with sodium channel blockers, even though many of them had already been exposed to this class of ASM. It has been shown that certain DS patients may respond to sodium channel blockers, especially LTG, with a greater frequency of seizures noted upon halting the medication.[32]
Prior to the age of 20, photosensitivity and pattern sensitivity each had a tendency to vanish, nevertheless some individuals still displayed light sensitivity.65 Consequently, for those who are older, triggering variables ought to be minimized.[32]
Prognosis
Numerous research have been performed to evaluate the DS prognosis. According to two studies, status epilepticus and sudden unexpected death in epilepsy (SUDEP) are the two most frequent causes of premature fatality among DS patients. Between ten and twenty percent of people with DS are thought to pass away before turning into 10 years of age. The International Dravet Syndrome Epilepsy Action League (IDEA League) conducted a study in which they concluded that 31/833 DS patients passed away within 10 years. The average death age was 4.6 years, with 19/31 deaths because of SUDEP, 10 from status epilepticus, 1 from ketoacidosis, and 1 from an accident. It is unclear what duration generally these patients are projected to live. A prospective study of 37 individuals showed that, by reducing status epilepticus from occurring at a young age, the prognosis for seizures and mental impairment in DS patients can be improved.[33]
Epidemiology
Dravet syndrome is a severe form of epilepsy, responsible for roughly 10% of cases in children.[34] It is a rare genetic disorder that affects an estimated 1 in every 20,000–40,000 births.[35][36]
History
Charlotte Dravet first described severe myoclonic epilepsy of infancy in Centre Saint Paul, Marseille, France in 1978 and the name was later changed to Dravet syndrome in 1989.[37] Similar descriptions were given by Bernardo Dalla Bernardina in Verona.[38]
Charlotte Figi, who was diagnosed as having Dravet syndrome, was the focus of a cause célèbre to provide a means for use of cannabidiol for persons with intractable seizures. She died from pneumonia, possibly caused by COVID-19, in April, 2020.[39]
References
- 1 2 3 4 5 "Dravet syndrome". www.epilepsydiagnosis.org. Archived from the original on 13 December 2022. Retrieved 9 September 2023.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 "Dravet Syndrome - Symptoms, Causes, Treatment | NORD". rarediseases.org. Archived from the original on 20 December 2022. Retrieved 9 September 2023.
- 1 2 3 Shorvon SD, Guerrini R, Cook M, eds. (2013). Oxford textbook of epilepsy and epileptic seizures. Oxford: Oxford Univ. Press. p. 13. ISBN 978-0-19-965904-3. Archived from the original on 2020-01-28. Retrieved 2022-06-25.
- 1 2 3 4 5 "Dravet Syndrome". National Institute of Neurological Disorders and Stroke. Archived from the original on 24 July 2023. Retrieved 9 September 2023.
- 1 2 "Dravet syndrome - About the Disease - Genetic and Rare Diseases Information Center". rarediseases.info.nih.gov. Archived from the original on 10 June 2023. Retrieved 9 September 2023.
- ↑ "Dravet syndrome". www.epilepsydiagnosis.org. Archived from the original on 11 August 2022. Retrieved 9 September 2023.
- ↑ "Dravet syndrome". www.epilepsydiagnosis.org. Archived from the original on 11 August 2022. Retrieved 9 September 2023.
- ↑ "Dravet syndrome". www.epilepsydiagnosis.org. Archived from the original on 11 August 2022. Retrieved 9 September 2023.
- ↑ Millichap, JJ; Koh, S; Laux, LC; Nordli DR, Jr (29 September 2009). "Child Neurology: Dravet syndrome: when to suspect the diagnosis". Neurology. 73 (13): e59-62. doi:10.1212/WNL.0b013e3181b9c880. PMID 19786689.
- ↑ Dravet, Charlotte; Guerrini, Renzo (18 September 2013). Dravet syndrome. John Libbey Eurotext. ISBN 978-2-7420-1231-2. Archived from the original on 10 September 2023. Retrieved 9 September 2023.
- 1 2 3 Selmer KK, Eriksson AS, Brandal K, Egeland T, Tallaksen C, Undlien DE (October 2009). "Parental SCN1A mutation mosaicism in familial Dravet syndrome". Clinical Genetics. 76 (4): 398–403. doi:10.1111/j.1399-0004.2009.01208.x. PMID 19673951. S2CID 40396775.
- 1 2 3 4 5 6 Granata T (April 2011). "Comprehensive care of children with Dravet syndrome". Epilepsia. 52 Suppl 2: 90–4. doi:10.1111/j.1528-1167.2011.03011.x. PMID 21463289. S2CID 573375.
- 1 2 3 4 Miller IO, de Menezes MA (April 2019). "SCN1A seizure disorders.". GeneReviews®[Internet]. Seattle: University of Washington. Archived from the original on 2020-10-27. Retrieved 2022-06-25.
- ↑ Aledo-Serrano Á, Mingorance A, Jiménez-Huete A, Toledano R, García-Morales I, Anciones C, Gil-Nagel A (May 2020). "Genetic epilepsies and COVID-19 pandemic: Lessons from the caregiver perspective". Epilepsia. 61 (6): 1312–1314. doi:10.1111/epi.16537. PMC 7276740. PMID 32420620.
- 1 2 3 Cheah C, Catterall WA (2012). "Characterizing the role of sodium channels in mouse models of Dravet Syndrome".
{{cite journal}}: Cite journal requires|journal=(help) - ↑ Couzin-Frankel J (8 April 2015). "Sudden death in epilepsy: Researchers finger possible cause". Science. doi:10.1126/science.aab2456.
- ↑ Lin YJ, Wen CN, Lin YY, Hsieh WC, Chang CC, Chen YH, et al. (January 2020). "Oil-in-water emulsion adjuvants for pediatric influenza vaccines: a systematic review and meta-analysis". Nature Communications. 11 (1): 315. Bibcode:2020NatCo..11..315L. doi:10.1038/s41467-019-14230-x. PMC 6965081. PMID 31949137.
- ↑ "What is Dravet Syndrome?". Cherry Hill, NJ: Dravet Syndrome Foundation, Inc. Archived from the original on 2016-10-28. Retrieved 2022-06-25.
- ↑ Ben-Menachem E (July 2011). "Vaccination and the onset of dravet syndrome". Epilepsy Currents. 11 (4): 120–2. doi:10.5698/1535-7511-11.4.120. PMC 3152151. PMID 21852883.
- 1 2 Wallace R (2005). "A plethora of SCN1A mutations: what can they tell us?". Epilepsy Currents. 5 (1): 17–20. doi:10.1111/j.1535-7597.2005.05105.x. PMC 1176321. PMID 16059449.
- ↑ Sugawara T, Tsurubuchi Y, Agarwala KL, Ito M, Fukuma G, Mazaki-Miyazaki E, et al. (May 2001). "A missense mutation of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction". Proceedings of the National Academy of Sciences of the United States of America. 98 (11): 6384–9. Bibcode:2001PNAS...98.6384S. doi:10.1073/pnas.111065098. PMC 33477. PMID 11371648.
- ↑ "Diagnostic Criteria". Dravet Syndrome Foundation. Archived from the original on 17 March 2015. Retrieved 17 March 2015.
- ↑ "Dravet Syndrome Foundation: Treatment". Archived from the original on 28 December 2015. Retrieved 1 January 2016.
- ↑ "NICE: Epilepsies: diagnosis and management". The National Institute for Health and Care Excellence (NICE). Archived from the original on 22 April 2019. Retrieved 1 January 2016.
- 1 2 3 4 5 Chiron C, Dulac O (April 2011). "The pharmacologic treatment of Dravet syndrome". Epilepsia. 52 Suppl 2: 72–5. doi:10.1111/j.1528-1167.2011.03007.x. PMID 21463285. S2CID 39978156.
- ↑ Brigo, Francesco; Igwe, Stanley C.; Bragazzi, Nicola Luigi (26 May 2020). "Stiripentol add-on therapy for drug-resistant focal epilepsy". The Cochrane Database of Systematic Reviews. 5: CD009887. doi:10.1002/14651858.CD009887.pub5. ISSN 1469-493X. PMC 7386790. PMID 32468572.
- ↑ "Press Announcements - FDA approves first drug comprised of an active ingredient derived from marijuana to treat rare, severe forms of epilepsy". www.fda.gov. Archived from the original on 23 April 2019. Retrieved 27 June 2018.
- ↑ Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, et al. (May 2017). "Trial of Cannabidiol for Drug-Resistant Seizures in the Dravet Syndrome" (PDF). The New England Journal of Medicine. 376 (21): 2011–2020. doi:10.1056/NEJMoa1611618. hdl:20.500.11820/2ed5e0db-7de9-4411-93d7-3a5c388006b9. PMID 28538134. S2CID 86481485. Archived (PDF) from the original on 2021-04-20. Retrieved 2022-06-25.
- ↑ Fintepla Archived 2022-01-20 at the Wayback Machine, on ema.europa.eu
- ↑ Fintepla (fenfluramine). An overview of Fintepla and why it is authorised in the EU Archived 2021-11-05 at the Wayback Machine, on ema.europa.eu
- ↑ FDA Approves New Therapy for Dravet Syndrome Archived 2022-04-29 at the Wayback Machine, on fda.gov
- 1 2 Cardenal-Muñoz E, Auvin S, Villanueva V, Cross JH, Zuberi SM, Lagae L, Aibar JÁ (March 2022). "Guidance on Dravet syndrome from infant to adult care: Road map for treatment planning in Europe". Epilepsia Open. 7 (1): 11–26. doi:10.1002/epi4.12569. PMC 8886070. PMID 34882995.
- ↑ Anwar A, Saleem S, Patel UK, Arumaithurai K, Malik P (June 2019). "Dravet Syndrome: An Overview". Cureus. 11 (6): e5006. doi:10.7759/cureus.5006. PMC 6713249. PMID 31497436.
- ↑ Berkovic SF (December 2020). "Epileptic encephalopathies of infancy: welcome advances". Lancet. 394 (10216): 2203–2204. doi:10.1016/S0140-6736(19)31239-5. PMID 31862247. S2CID 209394550.
- ↑ Hurst DL (August 1990). "Epidemiology of severe myoclonic epilepsy of infancy". Epilepsia. 31 (4): 397–400. doi:10.1111/j.1528-1157.1990.tb05494.x. PMID 1695145. S2CID 31868578.
- ↑ Yakoub M, Dulac O, Jambaqué I, Chiron C, Plouin P (September 1992). "Early diagnosis of severe myoclonic epilepsy in infancy". Brain & Development. 14 (5): 299–303. doi:10.1093/brain/aws151. PMID 1456383.
- ↑ Dravet C (April 2011). "The core Dravet syndrome phenotype". Epilepsia. 52 Suppl 2: 3–9. doi:10.1111/j.1528-1167.2011.02994.x. PMID 21463272. S2CID 41553756.
- ↑ "Bernardo Dalla Bernardina | University of Verona (UNIVR)". ResearchGate. Archived from the original on 2018-09-06. Retrieved 2022-06-25.
- ↑ Ingold J (April 8, 2020). "Charlotte Fiji". The Colorado Sun. Archived from the original on April 12, 2020. Retrieved April 13, 2020.
Charlotte Figi, the Colorado Springs girl who, as a gleeful and fragile child, launched a movement that led to sweeping changes in marijuana laws across the globe, has died from complications possibly related to the new coronavirus.
External links
| Classification | |
|---|---|
| External resources |