Unverricht–Lundborg disease
| Unverricht–Lundborg disease | |
|---|---|
| Other names: Progressive myoclonic epilepsy type 1[1] | |
| Specialty | Neurology |
| Frequency | Lua error in Module:PrevalenceData at line 5: attempt to index field 'wikibase' (a nil value). |
Unverricht–Lundborg disease (abbreviated ULD or EPM1) is the most common form of an uncommon group of genetic epilepsy disorders called the progressive myoclonus epilepsies.[2] It is caused due to a mutation in the cystatin B gene (CSTB).[3] The disease is named after Heinrich Unverricht, who first described it in 1891,[4][5] and Herman Bernhard Lundborg, who researched it in greater detail in 1901[6] and 1903.[7] ULD onsets in children between the ages of 6 and 16; there are no known cases in which the person was older than 18.[8] Most cases originate from the Baltic region of Europe, though many have been reported from countries in the Mediterranean.[3]
Onset of the disease is characterized by myoclonic jerks and tonic-clonic seizures.[8] Early cases often resulted in the need of a wheelchair and death before the age of 24,[9] but new treatments and medications have increased the life expectancy of individuals with ULD, in some cases even to near that of an unaffected individual.[8]
Signs and symptoms
Patients with Unverricht–Lundborg disease exhibit myoclonic jerks and tonic-clonic seizures at a young age, between ages 6–16. The myoclonic jerks occur in the muscles of the arms and legs closest to the torso, and are triggered due to a variety of common external stimuli.[8] Seizures begin at an average age of 10.8 years, with myoclonus beginning around 12.1 years.[9] It is not currently possible to diagnose without a genetic test, and since early symptoms are general, it is often mistaken for another more common epilepsy, in many cases juvenile myoclonic epilepsy (JME).[8]
Causes
The genetic cause of ULD is known, but research has led to new areas of study that may lead to an increase in knowledge of what causes ULD.
Genetic factors
The cause of ULD is known to be a mutation of the gene that produces cystatin B.[3] The disease is autosomal recessive, so both parents of an individual must be carriers of the recessive CSTB gene for the individual to inherit it, and for an individual to show symptoms of ULD, they must have both recessive CSTB genes.[3] Siblings of affected individuals who only have one recessive gene have been monitored and generally do not show the signs of ULD, though in some cases mild symptoms may be present.[10]
New developments
New research shows that cystatin B may not be the only factor involved in Unverricht–Lundborg disease. In a study, it was determined that patients with ULD had more dopamine receptors in certain areas of their brain than unaffected individuals.[11] The researchers chose to investigate dopamine receptors because they are known to be a factor in myoclonus, which are a significant part of the symptoms of ULD. The results of this study indicate that the cause of ULD may be more complex than currently thought.
Mechanism
While the genetic cause of Unverricht–Lundborg disease is known, the mechanism by which it works is not fully known. Current research has provided promising results that may lead to a confirmation of the mechanism. This research has been performed on mice with the gene for producing cystatin B removed, to provide a similar set of symptoms to individuals with ULD.[12] The mechanism currently supported by research is very similar to another theory of epilepsy progression known as kindling.[13]
Onset
Current research links cystatin B to production of inhibitory neurons known as GABAergic neurons. It has shown that a lack of cystatin B due to a mutation of the CSTB gene leads to a decrease in the number of inhibitory neurons, and this lack of inhibition makes the cells in the brain, particularly the hippocampus, more excitable. It is hypothesized that this increase in excitability is what causes the myoclonic jerks and tonic-clonic seizures in patients with ULD.[12]
Progression
Research also gives evidence to support the idea that cystatin B may be a type of "protecting" molecule in the brain. Normally, after a seizure, the presence of cystatin B prevents the neurons from dying due to toxic levels of neurotransmitters. Studies suggest that the absence of cystatin B leads to the death of affected neurons, leading to a damaged portion of the brain. This damage coupled with the increased excitability of the cells then leads to more damage, which is what makes Unverricht–Lundborg disease progressive.[12]
Diagnosis
The only currently available method to diagnose Unverricht–Lundborg disease is a genetic test to check for the presence of the mutated cystatin B gene. If this gene is present in an individual suspected of having the disease, it can be confirmed. However, genetic tests of this type are prohibitively expensive to perform, especially due to the rarity of ULD.[13] The early symptoms of ULD are general and in many cases similar to other more common epilepsies, such as juvenile myoclonic epilepsy.[8] For these reasons, ULD is generally one of the last options doctors explore when looking to diagnose patients exhibiting its symptoms. In most cases, a misdiagnosis is not detrimental to the patient, because many of the same medications are used to treat both ULD and whatever type of epilepsy the patient has been misdiagnosed with. However, there are a few epilepsy medications that increase the incidence of seizures and myoclonic jerks in patients with ULD, which can lead to an increase in the speed of progression, including phenytoin, fosphenytoin, sodium channel blockers, GABAergic drugs, gabapentin and pregabalin.[8]
Other methods to diagnose Unverricht–Lundborg disease are currently being explored. While electroencephalogram (EEG) is useful in identifying or diagnosing other forms of epilepsy, the location of seizures in ULD is currently known to be generalized across the entire brain. Without a specific region to pinpoint, it is difficult to accurately distinguish an EEG reading from an individual with ULD from an individual with another type of epilepsy characterized by generalized brain seizures. However, with recent research linking ULD brain damage to the hippocampus,[12] the usefulness of EEG as a diagnostic tool may increase.
Magnetic Resonance Imaging (MRI) is also often used during diagnosis of patients with epilepsy. While MRIs taken during the onset of the disease are generally similar to those of individuals without ULD, MRIs taken once the disease has progressed show characteristic damage[8]
While ULD is a rare disease, the lack of well defined cases to study and the difficulty in confirming diagnosis provide strong evidence that this disease is likely under diagnosed.[3]
Classification
Unverricht–Lundborg disease is also known as EPM1, as it is a form of progressive myoclonic epilepsy (PME). Other progressive myoclonic epilepsies include myoclonus epilepsy and ragged red fibers (MERRF syndrome), Lafora disease (EPM2a or EMP2b), Neuronal ceroid lipofuscinosis (NCL) and sialidosis. Progressive myoclonic epilepsies generally constitute only a small percentage of epilepsy cases seen, and ULD is the most common form. While ULD can lead to an early death, it is considered to be the least severe form of progressive myoclonic epilepsy.
Treatment
While there is no current cure to repair the mutated CSTB gene, several antiepileptic drugs are effective in reducing seizures and helping patients with ULD to manage the symptoms. In addition, new research is being performed to examine the effectiveness of other types of treatments.
Current methods
Valproic acid is the first line drug choice for reducing generalised seizures and myoclonus. Levetiracetam is also effective for both generalised seizures and myoclonus. Clonazepam and high-dose piracetam can alleviate myoclonus. Phenytoin can worsen seizures and may speed up neurodegeneration; carbamazepine, oxcarbazepine, tiagabine, vigabatrin, gabapentin and pregabalin may worsen myoclonus and myoclonic seizures.[14] Other common medications to treat ULD include topiramate and zonisamide. If an individual with Unverricht–Lundborg disease is particularly sensitive to a certain type of stimulus, it is also beneficial to reduce the patient's exposure to that stimulus in order to reduce the likelihood of seizures.[8] Since ULD is progressive and may not get better over time, depression has been documented in many cases, so providing a strong support group of friends, family, and even other individuals with ULD is very beneficial.[9]
Prognosis
For early Unverricht–Lundborg disease patients, the disease would begin to progress early and lack of effective treatment meant a quick progression. In many cases the patient would require a wheelchair for mobility, and would die at a young age.[9]
However, increased knowledge about the disease and improved treatment and medication has led to a dramatic improvement in prognosis for individuals with ULD. Antiepileptic drugs reduce the occurrence of seizures and myoclonus, which leads to a decrease in the damage caused in the brain due to seizures and the body due to falls resulting from the seizures. As a result, individuals with Unverricht–Lundborg disease are now much less likely to end up in a wheelchair, which eliminates the chance of complications involved with being a wheelchair user.[13] All these factors have increased the outlook for patients. Due to the progressive nature of the disease, depression is prevalent,[9] but support of family and friends as well as proper treatment can help. While early patients with ULD had a life expectancy of around 24 years,[9] there have recently been reported cases of individuals living to near-normal ages.[8]
Epidemiology
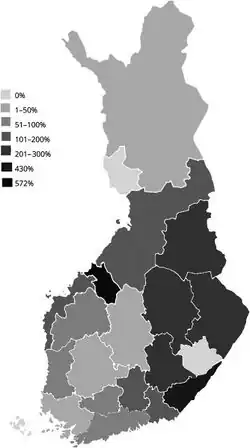
The only country that Unverricht–Lundborg disease has a reported incidence is in Finland, where it is reported to occur in 4 in 100,000 individuals.[8]
However, ULD has only become well defined recently, and it is likely still under diagnosed,[3] so the actual incidence may be different that what is currently known. Other countries with known cases include countries in the Mediterranean region including Italy, France, Tunisia, Algeria, and Morocco,[8] as well as the United States.
History
Unverricht–Lundborg disease was first known as one of two different diseases, depending on the location of the individual who had it: Baltic myoclonus or Mediterranean myoclonus.[8] The reason for the different names was partly regional but also because the prognosis of the disease was different for individuals with each due to the way that it was treated in that region. Eventually, both were realized to be the same disease, ULD.
Research directions
Many studies have been performed recently to investigate the cause, mechanism, and chemical basis of Unverricht–Lundborg disease.
Cystatin B Characteristics
A recent study has attempted to describe the behavior of normal and mutated cystatin B as it is expressed in the body. The results show that cystatin B has a polymeric structure, and that the mutated form of cystatin B, which is present in patients with Unverricht–Lundborg disease, is likely to attract other molecules of cystatin B and form clumps of the molecule. The researchers suggest that this clotting action of the cystatin B molecules may be one of the factors that cause progression of ULD.[15]
Study of Heterozygous Mice
In humans, it is generally known that unless a patient has both recessive CSTB genes (are homozygous recessive), they will not express ULD symptoms. A recent study has attempted to characterize the effects, if any, seen in mice that carry only one recessive CSTB gene (are heterozygous). The researchers analyzed normal and heterozygous mice by having them perform various tasks. The study found that heterozygous mice performed similar to normal mice when the task was started, but as the task continued or became more complex they were more likely to fail. While the results for the heterozygous mice were not remarkably different from the normal mice, they do indicate that carrying just one recessive CSTB gene may have adverse effects, at least in mice.[10]
Analysis of EEG as ULD Progresses
Currently, electroencephalography (EEG) is not very effective as a diagnostic tool for Unverricht–Lundborg disease. This study instead looks to characterize the change in EEG of ULD patients as the disease progresses. The researchers studied twenty-five patients with ULD and monitored their EEG over time. The results show that certain brain waves that are present at the beginning of ULD progression and are also present in unaffected individuals, including spontaneous generalized spike or polyspike wave discharges and photoparoxysmal response, tend to decrease after 10 to 15 years.[16]
References
- ↑ RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Progressive myoclonic epilepsy type 1". www.orpha.net. Archived from the original on 26 July 2023. Retrieved 21 October 2023.
- ↑ Saneto, Russell P (editor). Unverricht-Lundborg Disease Archived 2013-10-29 at the Wayback Machine. epilepsy.com.
- 1 2 3 4 5 6 Joensuu T, Lehesjoki AE, Kopra O. 2008. Molecular background of EPM1-Unverricht-Lundborg disease. Epilepsia 49:557-63
- ↑ Unverricht H. Ueber familiäre Myoklonie. Dtsch Z Nervenheilk 1891; 7: 32–67
- ↑ Unverricht H. Die Myoclonie. Leipzig und Wien, F. Deuticke 1891
- ↑ Lundborg H. Ueber Degeneration und degenerierte Geschlechter in Schweden. I. Klinische Studien und Erfahrungen hinsichtlich der familiären Myoklonie und damit verwandter Krankheiten. Stockholm, I. Marcus’ Boktr.-Aktiebolag 1901 (= Medizinische Dissertation, zur öffentlichen Verteidigung vorgelegt am 22.5.1901)
- ↑ Lundborg H. Die progressive Myoklonus-Epilepsie (Unverricht’s Myoklonie). Upsala, Almqvist und Wiksell’s Buchdruckerei 1903
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Kalviainen R, Khyuppenen J, Koskenkorva P, Eriksson K, Vanninen R, Mervaala E. 2008. Clinical picture of EPM1-Unverricht-Lundborg disease. Epilepsia 49:549-56
- 1 2 3 4 5 6 Chew NK, Mir P, Edwards MJ, Cordivari C, Martino D, et al. 2008. The natural history of Unverricht-Lundborg disease: A report of eight genetically proven cases. Movement Disorders 23:107-13
- 1 2 Kaasik A, Kuum M, Aonurm A, Kalda A, Vaarman A, Zharkovsky A. 2007. Seizures, Ataxia, and Neuronal Loss in Cystatin B Heterozygous Mice. Epilepsia 48(4):752-57
- ↑ Korja M, Kaasinen V, Lamusuo S, Parkkola R, Nagren K, Marttila R. 2007. Substantial Thalamostriatal Dopaminergic Defect in Unverricht-Lundborg Disease. Epilepsia 48(9):1763-73
- 1 2 3 4 Franceschetti S, Sancini G, Buzzi A, Zucchini S, Paradiso B, et al. 2007. A pathogenetic hypothesis of Unverricht-Lundborg disease onset and progression. Neurobiology of Disease 25:675-85
- 1 2 3 Warmouth G., Emory University Department of Epilepsy, interviewed by M. Scrudato, Nov. 19, 2008
- ↑ Kälviäinen, R.; Khyuppenen, J.; Koskenkorva, P.; Eriksson, K.; Vanninen, R.; Mervaala, E. (Apr 2008). "Clinical picture of EPM1-Unverricht-Lundborg disease". Epilepsia. 49 (4): 549–56. doi:10.1111/j.1528-1167.2008.01546.x. PMID 18325013.
- ↑ Cipollini E, Riccio M, Di Giaimo R, Dal Piaz F, Pulice G, Catania S, Caldarelli I, Dembic M, Santi S, Melli M. 2008. Cystatin B and its EPM1 mutants are polymeric and aggregate prone in vivo. Biochimica et Biophysica Acta 1783: 312-22
- ↑ Ferlazzo E, Magaudda A, Striano P, Vi-Hong N, Serra S, Genton P. 2007. Long-term evolution of EEG in Unverricht-Lundborg disease. Epilepsy Research 73:219-27
External links
- Genetics Home References Archived 2010-04-22 at the Wayback Machine
- GeneReviews Archived 2010-03-04 at the Wayback Machine
| Classification | |
|---|---|
| External resources |
|